library(mia)
# Import data and rename
data("HintikkaXOData")
mae <- HintikkaXOData
# Agglomerate rare taxa by prevalence at the Family level & log10-transform read counts
mae[["microbiota"]] <- agglomerateByPrevalence(mae[["microbiota"]], rank = "Family")
mae[["microbiota"]] <- transformAssay(mae[["microbiota"]], method = "clr", pseudocount = 1)
# Get cross-correlation between taxa & metabolites
x <- testExperimentCrossAssociation(mae, experiment1 = "microbiota", experiment2 = "metabolites",
assay.type1 = "clr", assay.type2 = "nmr",
mode = "matrix", sort = TRUE)$cor
# Visualize taxa-metabolite associations on heatmap
ComplexHeatmap::Heatmap(x)Orchestrating microbiome multi-omics
Bioconductor conference, 2023
Orchestrating microbiome multi-omics with R and Bioconductor
Leo Lahti @antagomir | Department of Computing, University of Turku, Finland

Organizing microbiome data
Side information
sample & feature metadata
Multiple assays
counts, relabundance, phILR..
Alternative experiments
16S, metagenome, phylogenetic microarrays; pipelines
Cross-kingdom analyses bacteriome, archaeome, virome, mykobiome, eukaryome
Hierarchical data
taxonomic levels, nested designs, sample & feature trees
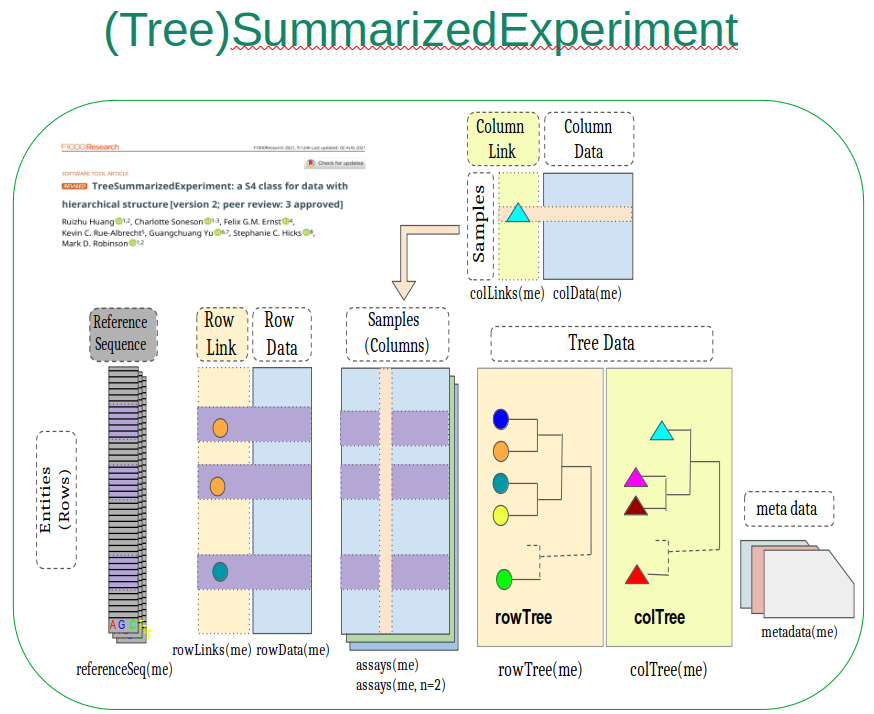
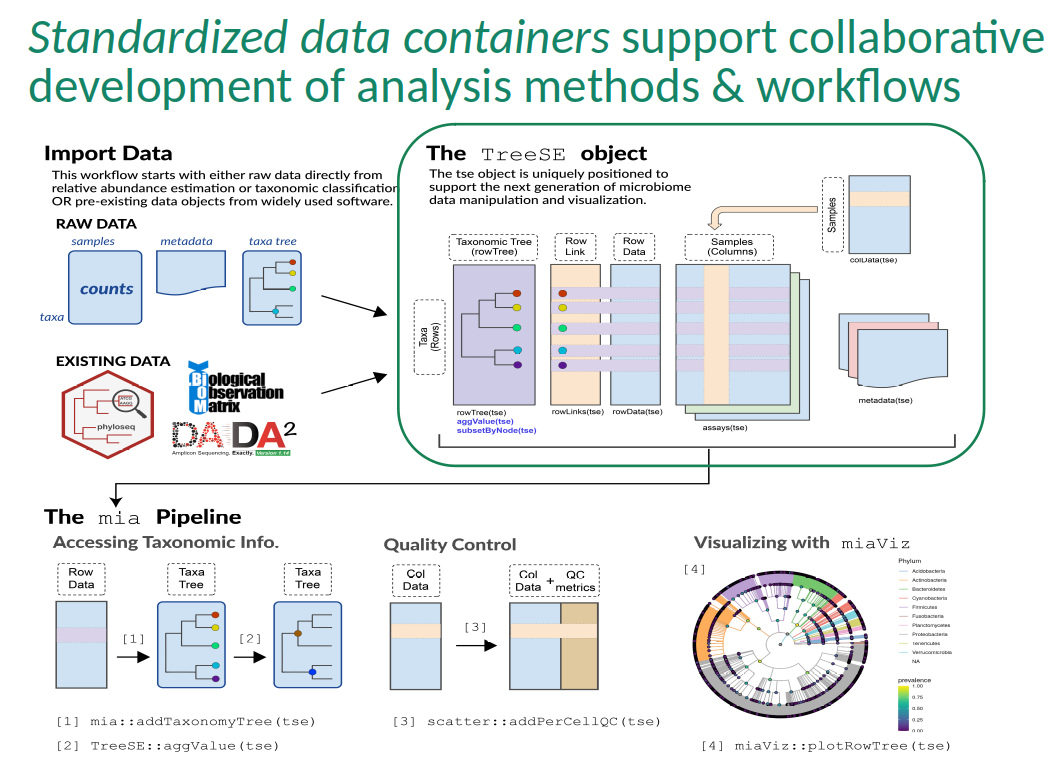
Fig. by Domenick Braccia (EuroBioC 2020)
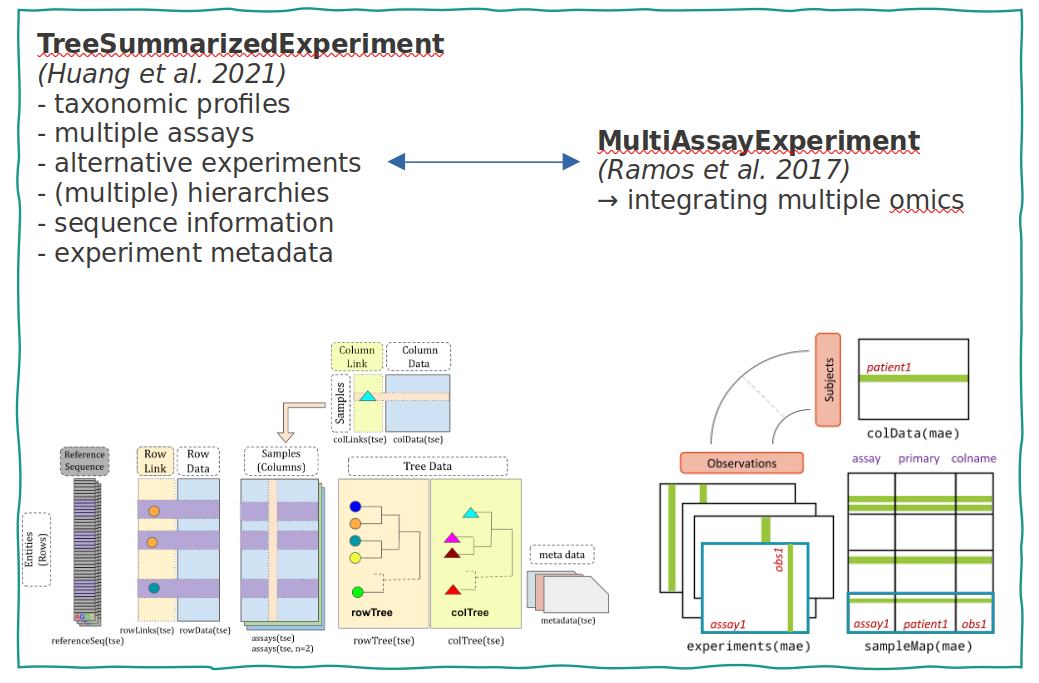
Integrating multiple experiments
Combine taxonomic, metagenomic & functional profiling, and host measurements
by extending the Bioconductor MultiAssayExperiment class.
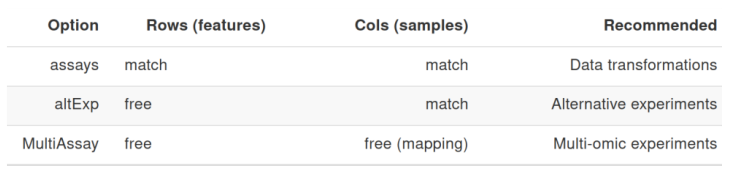

Benefits for microbiome data science
Extended
enhances multi-assay analyses in microbiome studies
Optimized
speed, memory & scale (sample sizes, datasets, cohorts)
Integrated
with other applications & frameworks (e.g. Single Cell, Spatial transcriptomics)
Package and data ecosystem
Bioconductor methods & packages supporting the framework.
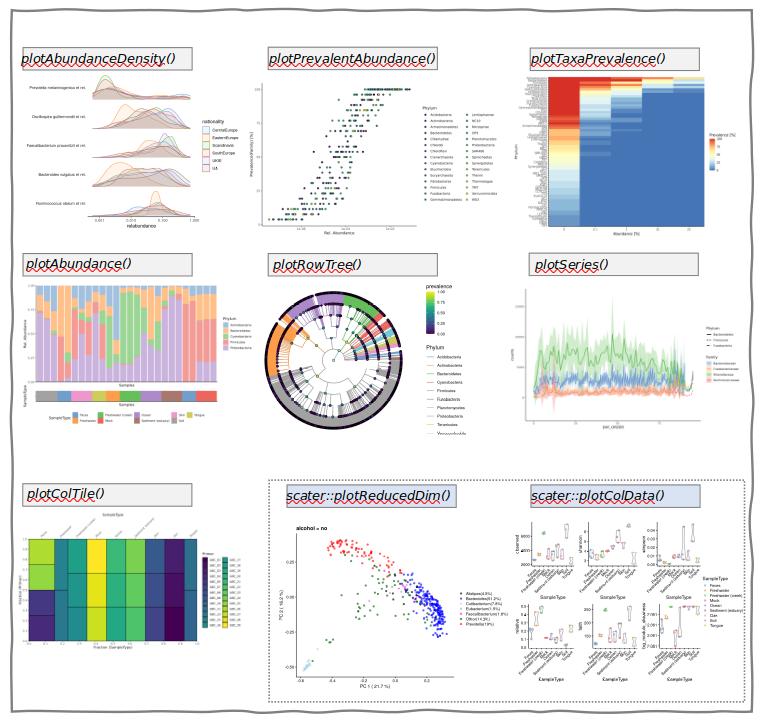
Basic wrangling: transformations, agglomeration, split/merge etc.
Alpha & beta diversity
Dimension reduction (scater)
Differential abundance (ANCOMBC, ALDEx2, benchdamic)
Visualization (miaViz)
Time series manipulation (miaTime)
Ecological simulations (miaSim)
![]()
![]()
Importers for other common formats
Linking with other common formats and microbiome data science frameworks.




Open data resources
Open microbiome data sets readily available in TreeSummarizedExperiment format via curatedMetagenomicData, EBI MGnify, Bioconductor ExperimentHub, and packages.


Minimal example
Cross-correlating taxonomic and metabolomic profiles from a dietary intervention study in mice (Hintikka et al. 2021).

Documentation: OMA Gitbook (beta)
Complements other Bioconductor Gitbooks; overlapping methods and analysis strategies: microbiome.github.io/OMA



Community
Coordination: Tuomas Borman, Leo Lahti
Giulio Benedetti, Yağmur Şimşek, Basil Courbayre, Jeba Akewak, Daena Rys, Henrik Eckermann, Chouaib Benchraka, Rajesh Shigdel, Artur Sannikov, Lu Yang, Renuka Potbhare, S. A. Shetty, R. Huang, F. G.M. Ernst, D. J. Braccia, H. C. Bravo; 20+ contributors, 10+ countries. Welcome to join!
Acknowledgments: TreeSummarizedExperiment, SingleCellExperiment, MultiAssayExperiment, curatedMetagenomicData, MGnifyR, phILR, ANCOMBC, ALDEx2, benchdamic, scater, scuttle authors..
Courses in 2023: Pune (IND) | Turku & Oulu (FIN) | Utrecht, Wageningen & Radboud (NLD) | Norwich (UK) |Bergen (NO)



![]()



![]()
![]()
Bioconductor books

Resources
Microbiome R package listing:
github.com/microsud/Tools-Microbiome-Analysis
![]()

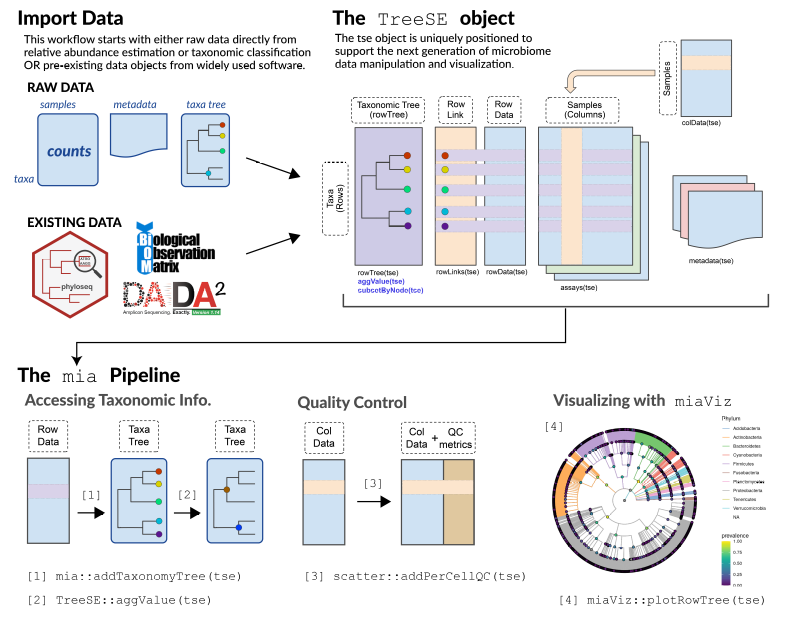
Microbiome data science workflow
Data containers:
TreeSummarizedExperiment
MultiAssayExperiment
R/Bioc packages:
mia, miaViz
Misc
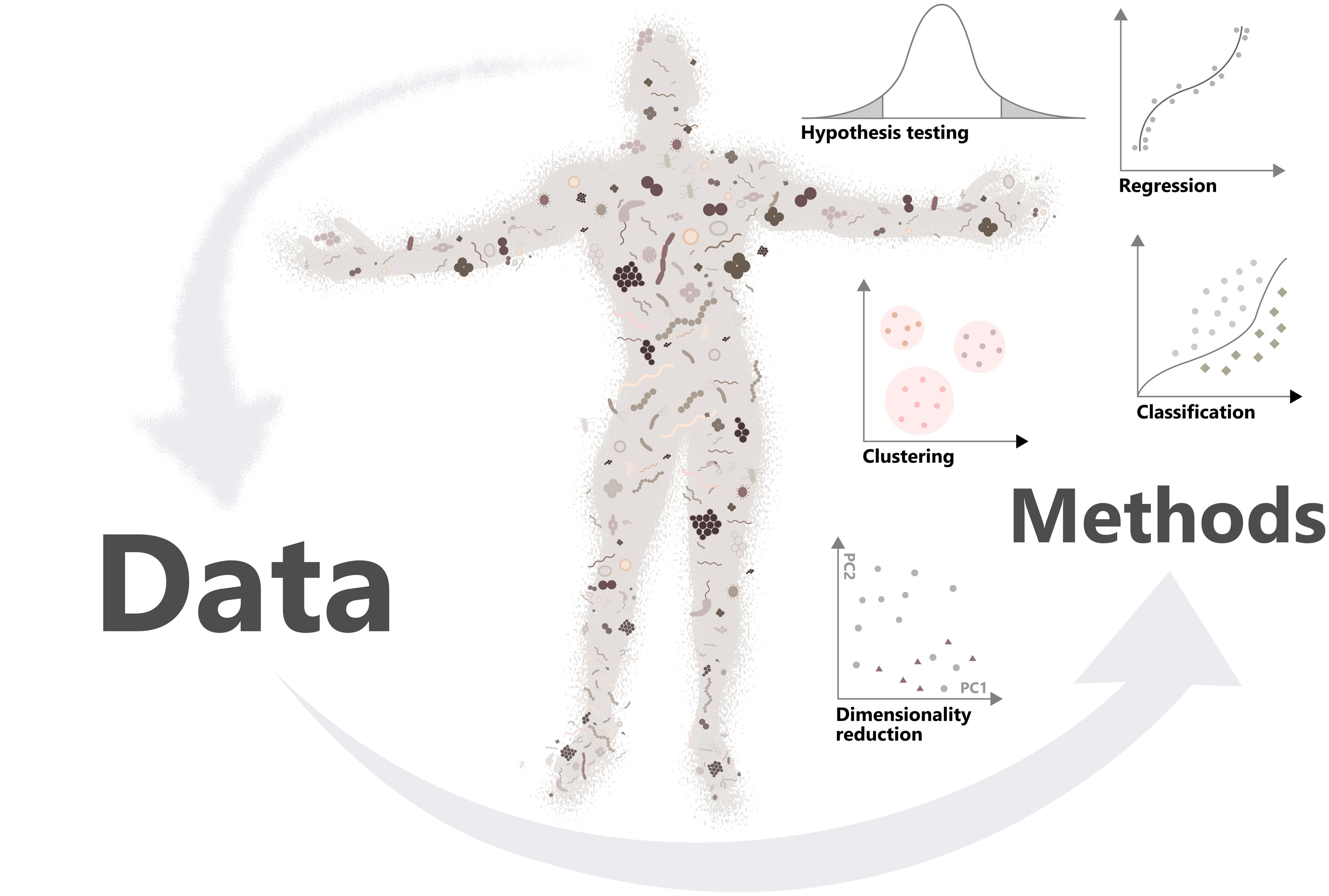
Figure source: Moreno-Indias et al. (2021) Statistical and Machine Learning Techniques in Human Microbiome Studies: Contemporary Challenges and Solutions. Frontiers in Microbiology 12:11.
TreeSummarizedExperiment
Optimal container for microbiome data integration?
Improve interoperability, ensure sustainability.
Multiple assays
seamless interlinking
Side information
extended capabilities & data types
Hierarchical data
both samples & features
Optimized
speed & memory
Integrated
other applications & frameworks

Optimal container for microbiome data integration?
Improve interoperability, ensure sustainability.
Multiple assays
seamless interlinking
Side information
extended capabilities & data types
Hierarchical data
both samples & features
Optimized
speed & memory
Integrated
other applications & frameworks
Linking multiple omics
Taxonomic, metagenomic & functional profiling, host measurements