Multivariate analysis
- Multiple variables
- Different methods
- Ordination-based methods
- Clustering
- Classification, …
Ordination
- Beta diversity: diversity between microbial communities
- Simplify and visualize high-dimensional data
- Projects data into lower dimensional latent space
![]()
Matrix factorization
- Decomposes complex data into components
- Widely used and general technique
- Methods vary based on goals and constraints
Ordination methods
- Different methods
- Euclidean/non-Euclidean
- Unsupervised/supervised
Principal component analysis (PCA)
- Goal: Maximize the variance
- Euclidean distance
- Aitchison distance: CLR + Euclidean distance
Example 1.1: PCA
First we load an example dataset and apply robust clr transformation.
Show code
library(mia)
data("Tengeler2020")
tse <- Tengeler2020
# Transform data
tse <- transformAssay(tse, method = "rclr")
Then we perform PCA with runPCA()function available in scater package.
Show code
library(scater)
tse <- runPCA(
tse,
assay.type = "rclr"
)
We can retrieve a list of all reduced dimensions with reducedDims().
Show code
List of length 1
names(1): PCA
If you need only their names, these can be accessed with reducedDimNames().
And the results can be accessed with reducedDim(tse, "PCA").
Show code
reducedDim(tse, "PCA") |> head()
PC1 PC2 PC3 PC4 PC5 PC6
A110 -4.5035339 -2.456547 -2.11328100 -3.8634688 2.0123949 -0.3325532
A12 2.4156363 4.982008 -0.07545471 0.1898264 -1.3925704 1.2237517
A15 -2.7404749 -3.121391 -4.46833577 8.1698047 -2.5837119 -2.1243919
A19 0.7358807 7.090486 -1.00797353 -1.2989823 0.1064523 -3.3824751
A21 0.4648846 5.448022 -1.14685965 -0.8743427 1.2143298 -4.4323282
A23 -2.0686346 -1.954092 -2.61287624 -2.5249816 -3.0007937 -2.4024629
PC7 PC8 PC9 PC10 PC11 PC12
A110 -0.8342411 1.0193914 -4.2216208 1.1932256 0.172707622 -2.3565927
A12 -0.3055218 0.7968376 0.4988079 2.7782003 0.551278116 0.4895880
A15 -1.3546507 1.5617929 -0.7468926 -0.7977925 -0.002285458 -1.9595096
A19 1.4979577 0.1788277 -0.9825262 -2.3851223 -2.018251650 -1.4132247
A21 1.6593740 0.5619416 0.6748281 -0.2531164 -1.974152848 0.7930157
A23 -2.5300350 -3.0190698 3.9427392 0.2632914 1.180388830 0.3831161
PC13 PC14 PC15 PC16 PC17 PC18
A110 0.2449630 -1.2664497 1.4364113 0.89583856 0.8365374 0.4457789
A12 -1.4948351 -2.5473097 -0.8648291 -1.48252017 -0.7839398 0.1476567
A15 0.4154040 -0.2455156 0.1621916 0.33095787 -0.7875945 -0.0824129
A19 0.1953649 0.9069244 0.2419303 0.38847386 1.4955734 0.3421782
A21 -1.2728108 0.7827710 -0.8452163 -0.58884502 -1.9694914 -0.5016079
A23 -0.8302655 -0.2890572 2.1834383 0.09925442 0.3728412 -1.4058642
PC19 PC20 PC21 PC22 PC23 PC24
A110 0.987010794 -1.0053150 -0.9899730 0.8829111 -0.2200736 -0.2975089
A12 0.114383997 0.5743782 -1.9702511 -0.4000117 -0.2989457 -1.0332134
A15 -0.517904963 0.1558602 -0.2901768 -0.1773831 -0.4550756 -0.1875868
A19 0.480395457 1.3345829 0.8691778 -0.5697885 0.2015948 -0.7871444
A21 0.006978105 -1.8347856 -0.6948813 0.4662380 -0.3698014 0.5050710
A23 0.516079026 -0.8406535 0.3192697 0.3128884 -0.1924474 -0.2425484
PC25 PC26
A110 -0.60890560 -0.20643929
A12 0.51342549 -1.06913557
A15 -0.04222125 0.01149364
A19 1.10441000 -0.77053190
A21 -1.05782664 0.59576805
A23 0.74942196 -0.01116033
Example 1.2: Visualize PCA
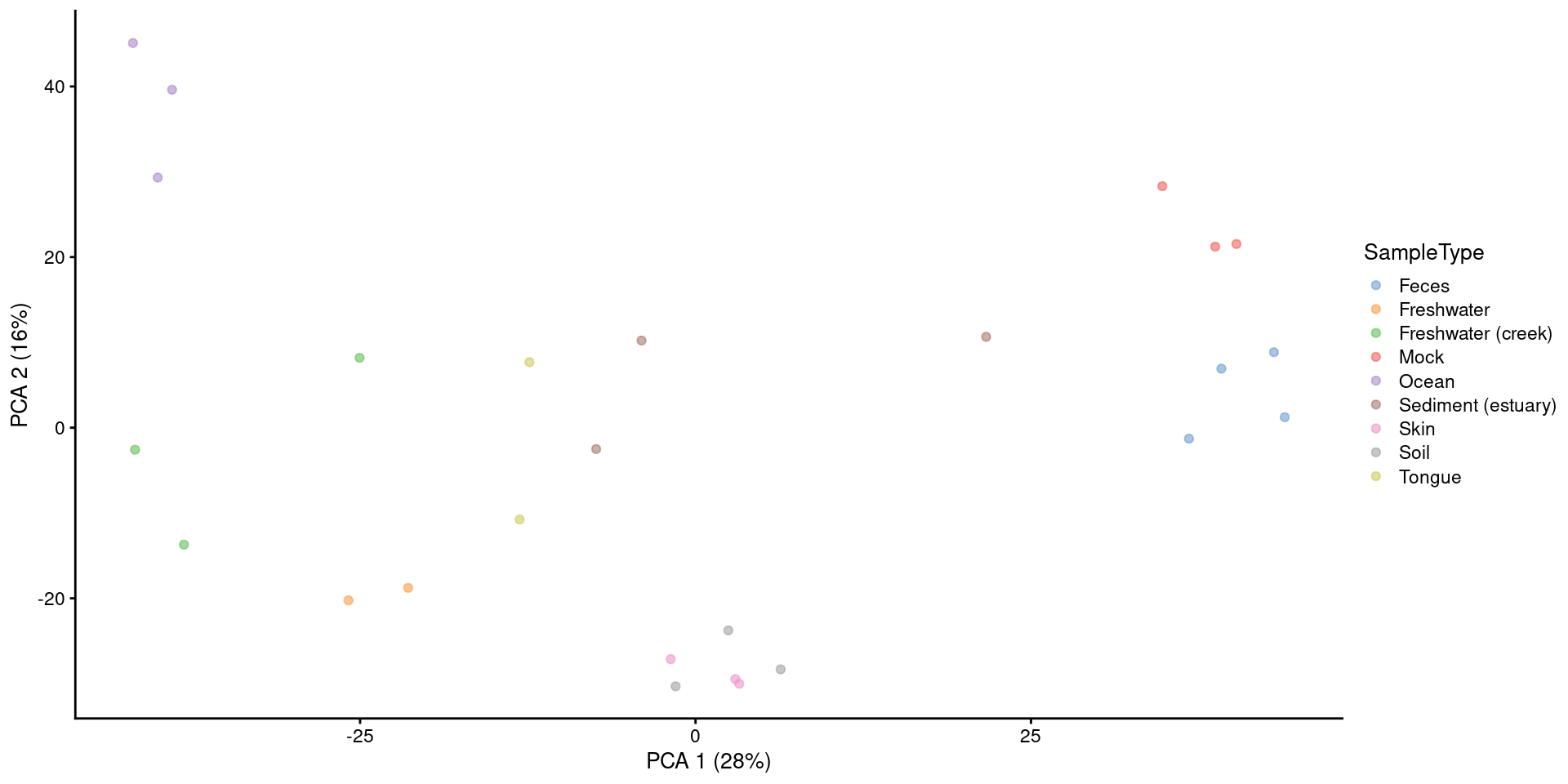
PCA or other ordination results are usually visualized with scatter plot.
Show code
plotReducedDim(
tse,
dimred = "PCA",
colour_by = "patient_status"
)
![]()
Example 1.3: PCA contributors
Some taxa contribute more than others to the generation of reduced dimensions.
Show code
library(miaViz)
plotLoadings(tse, "PCA", ncomponents = 2)
![]()
Exercises 1: PCA
- 29.7.1 Reduced dimensions retrieval
- 29.7.2 Visualization basics with PCA
Principal coordinate analysis (PCoA)
- Goal: Preserve the dissimilarity structure
- Non-Euclidean
- Different dissimilarity metrics
When Euclidean distance is used, PCoA reduces to PCA.
Example 2.1: PCoA
We transform the counts assay to relative abundances and store the new assay back into the TreeSE.
Show code
# Transform counts to relative abundance
tse <- transformAssay(tse, method = "relabundance")
Here, we run PCoA on the relative abundance assay to reduce the dimensionality of the data. We set method to Bray-Curtis dissimilarity.
Show code
# Run PCoA with Bray-Curtis dissimilarity
tse <- runMDS(
tse,
assay.type = "relabundance",
FUN = getDissimilarity,
method = "bray"
)
We can see that now there are additional results in reducedDim.
Example 2.2: Visualize PCoA
Similarly to PCA, we can visualize PCoA with scatter plot.
Show code
plotReducedDim(
tse,
dimred = "MDS",
colour_by = "patient_status"
)
![]()
Example 2.3: PCoA contributors
Loadings of PCoA cannot be interpreted as directly as PCA: “features that contribute the most to dissimilarity”.
Instead, we can calculate correlation between abundances and coordinates.
Show code
# Compute correlation between features and reduced dimensions
comp_loads <- apply(
assay(tse, "relabundance"),
MARGIN = 1, simplify = FALSE,
function(x) cor(x, reducedDim(tse, "MDS"), method = "kendall")
)
# Prepare matrix of feature loadings
taxa_loads <- do.call(rbind, comp_loads)
colnames(taxa_loads) <- paste0("MDS", seq(ncol(taxa_loads)))
rownames(taxa_loads) <- rownames(tse)
The top PCoA loadings for the first two dimensions are visualised below.
Show code
plotLoadings(taxa_loads, ncomponents = 2)
![]()
Exercises 2: PCoA
- 29.7.3 Principal Coordinate Analysis (PCoA)
Redundancy analysis (RDA)
- Supervised
- How much covariate explains the differences in microbial profile?
- Two steps
- Principal Coordinate Analysis (PCoA)
- Maximizes the variance explained by covariates
Example 2.1: dbRDA
We can apply dbRDA with runRDA() function. formula tells how the method is applied; here “patient_status” and cohort are the explanatory variables.
Show code
# Run PCoA with Bray-Curtis dissimilarity
tse <- addRDA(
tse,
formula = x ~ patient_status + cohort,
assay.type = "relabundance",
method = "bray"
)
Example 2.2.: Visualize dbRDA
We can visualize dbRDA results with plotReducedDim() or plotRDA() which have additional features.
Show code
plotRDA(
tse,
dimred = "RDA",
colour_by = "patient_status"
)
![]()
Exercises 3: dbRDA
- 29.7.5 Redundancy analysis (RDA)
- 29.7.6 Beta diversity analysis
Extra:
Find the top 5 contributor taxa for principal component 1.
Example 3.1: Other Distances
A different distance function can be specified with FUN, such as phylogenetic distance.
Show code
# Run PCoA with Unifrac distance
tse <- runMDS(
tse,
assay.type = "counts",
FUN = getDissimilarity,
method = "unifrac",
tree = rowTree(tse),
ncomponents = 3,
name = "Unifrac")
The number of dimensions to visualise can also be adjusted with ncomponents.
Show code
# Visualise Unifrac distance between samples
plotReducedDim(
tse,
"Unifrac",
ncomponents = 3,
colour_by = "patient_status"
)
![]()
Example 3.2: Comparison
Different ordination methods return considerably different results, which can be compared to achieve a better understanding of the data.
Show code
library(patchwork)
# Generate plots for
plots <- lapply(reducedDimNames(tse), function(name){
plotReducedDim(tse, name, colour_by = "patient_status")
})
# Generate multi-panel plot
wrap_plots(plots) +
plot_layout(guides = "collect") +
plot_annotation(tag_levels = "A")
![]()
Exercise 3
Run MDS on the CLR assay with Euclidean distance and compare the results with the previous PCoA and PCA.
Extra:
Make a plot with the first three dimensions, and a plot with the second and fourth dimensions.