Calculation of Double Principal Correspondence analysis
Source:R/AllGenerics.R, R/runDPCoA.R
runDPCoA.RdDouble Principal Correspondence analysis is made available via the
ade4 package in typical fashion. Results are stored in the
reducedDims and are available for all the expected functions.
getDPCoA(x, y, ...)
# S4 method for class 'ANY,ANY'
getDPCoA(
x,
y,
ncomponents = 2,
ntop = NULL,
subset.row = subset_row,
subset_row = NULL,
scale = FALSE,
transposed = FALSE,
...
)
# S4 method for class 'TreeSummarizedExperiment,missing'
getDPCoA(
x,
...,
assay.type = assay_name,
assay_name = exprs_values,
exprs_values = "counts",
tree.name = tree_name,
tree_name = "phylo"
)
calculateDPCoA(x, ...)
addDPCoA(x, ..., altexp = NULL, name = "DPCoA")
runDPCoA(x, ...)Arguments
- x
- y
a
distor a symmetricmatrixcompatible withade4:dpcoa- ...
Currently not used.
- ncomponents
Numeric scalar. Indicates the number of DPCoA dimensions to obtain. (Default:2)- ntop
Numeric scalar. Specifies the number of features with the highest variances to use for dimensionality reduction. AlternativelyNULL, if all features should be used. (Default:NULL)- subset.row
Character Vector. Specifies the subset of features to use for dimensionality reduction. This can be a character vector of row names, an integer vector of row indices or a logical vector. (Default:NULL)- subset_row
Deprecated. Use
subset.rowinstead.- scale
Logical scalar. Should the expression values be standardized? (Default:FALSE)- transposed
Logical scalar. Specifies if x is transposed with cells in rows. (Default:FALSE)- assay.type
Character scalar. Specifies the name of assay used in calculation. (Default:"counts")- assay_name
Deprecated. Use
assay.typeinstead.- exprs_values
Deprecated. Use
assay.typeinstead.- tree.name
Character scalar. Specifies the name of the tree to be included in the phyloseq object that is created, (Default:"phylo")- tree_name
Deprecated. Use
tree.nameinstead.- altexp
Character scalarorinteger scalar. Specifies an alternative experiment containing the input data. (Default:NULL)- name
Character scalar. A name for the column of thecolDatawhere results will be stored. (Default:"DPCoA")
Value
For getDPCoA a matrix with samples as rows and CCA dimensions as
columns
For addDPCoA a modified x with the results stored in
reducedDim as the given name
Details
For addDPCoA a
TreeSummarizedExperiment containing
the expression values as well as a rowTree to calculate y using
cophenetic.phylo.
In addition to the reduced dimension on the features, the reduced dimension
for samples are returned as well as sample_red attribute.
eig, feature_weights and sample_weights are
returned as attributes as well.
See also
Examples
data(esophagus)
dpcoa <- getDPCoA(esophagus)
head(dpcoa)
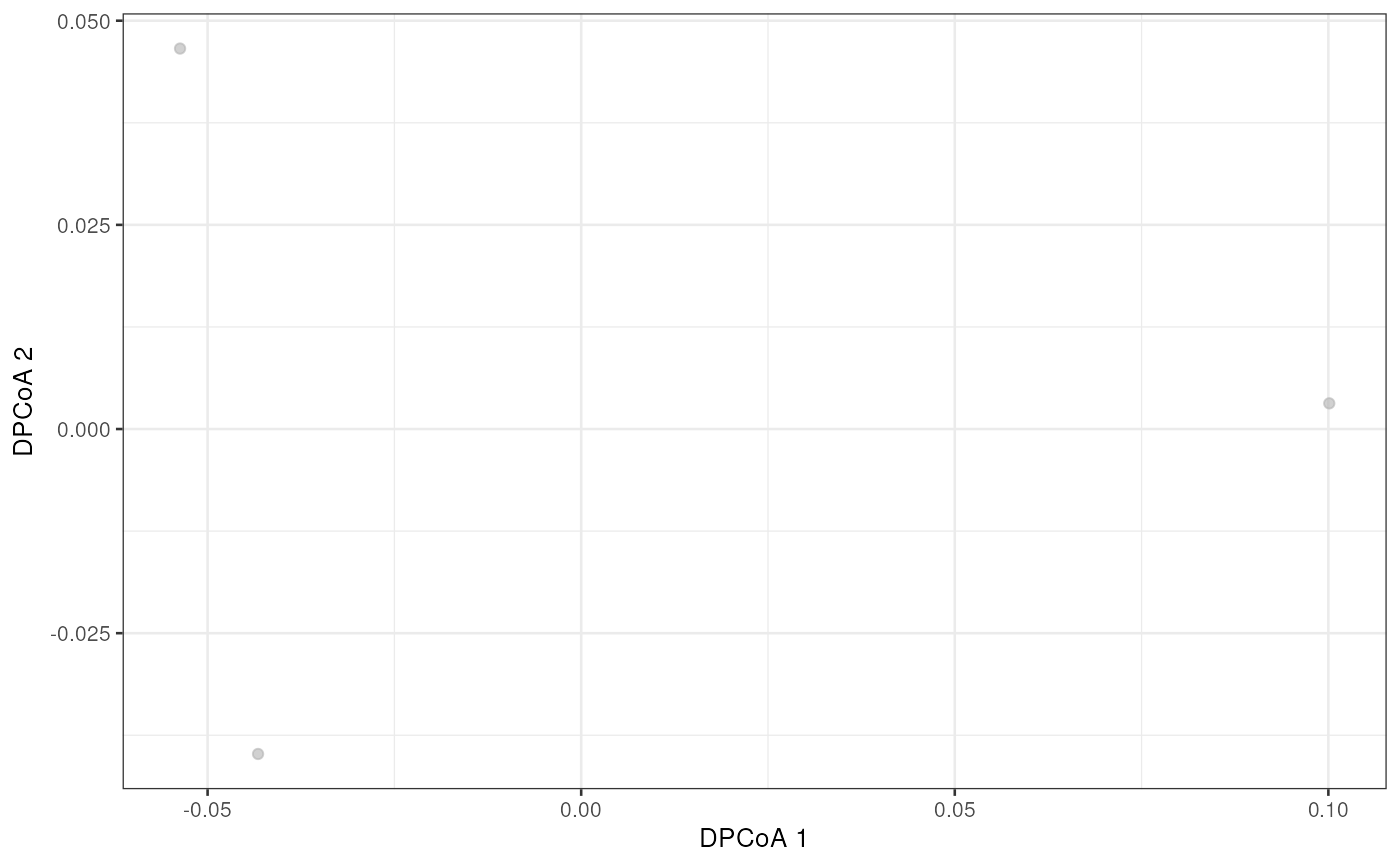
#> [,1] [,2]
#> B -0.05368615 0.046589779
#> C -0.04324591 -0.039790060
#> D 0.10011870 0.003144932
esophagus <- addDPCoA(esophagus)
reducedDims(esophagus)
#> List of length 1
#> names(1): DPCoA
library(scater)
plotReducedDim(esophagus, "DPCoA")