2 Microbiome Data
2.1 Data science framework
The building blocks of the framework are data containers (SummarizedExperiment and its derivatives), packages from various developers using the TreeSE container, open demonstration datasets, in a separate chapter Section 2.3, and online tutorials, including this online book as well as the various package vignettes and other materials.
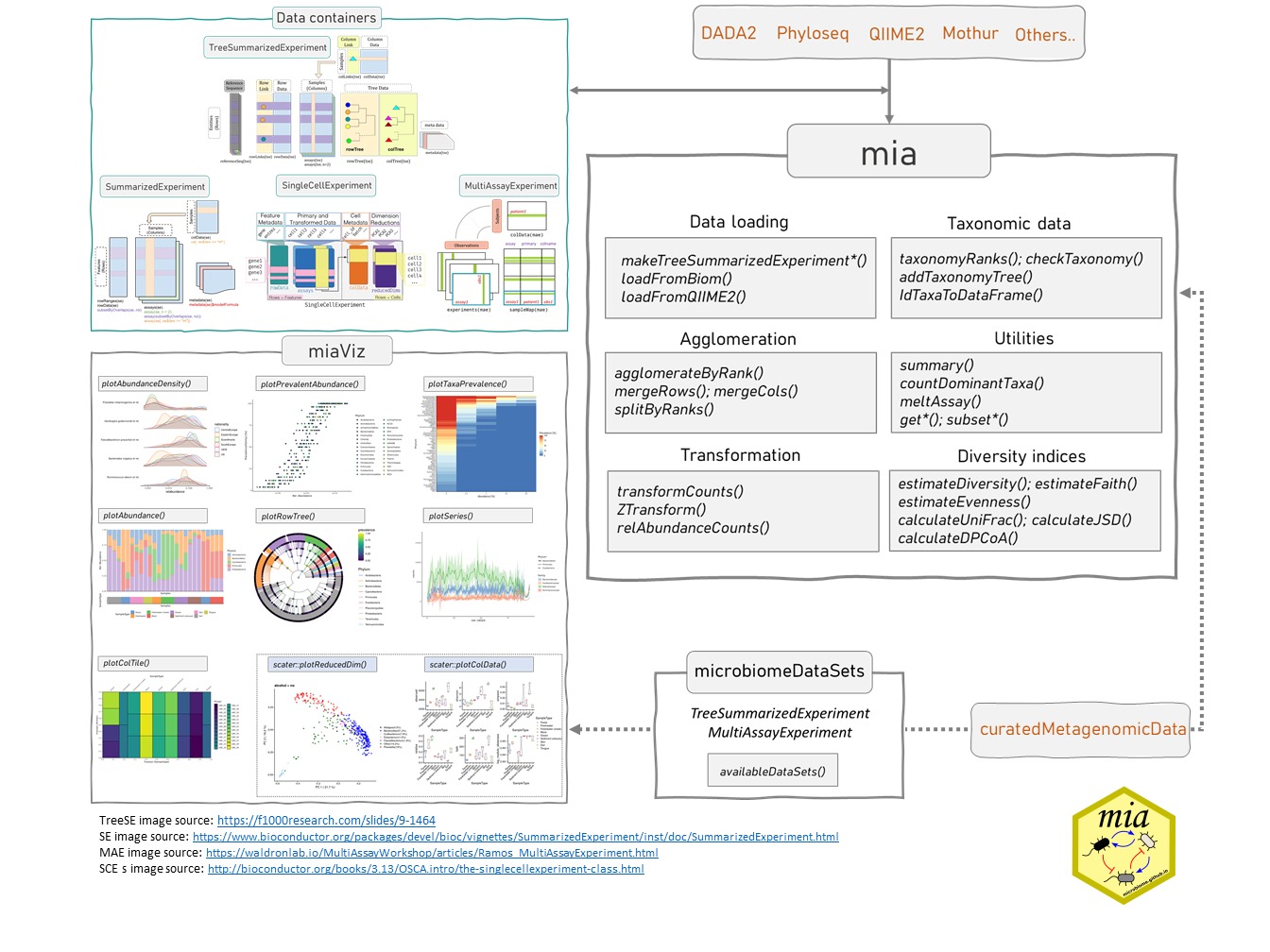
2.2 Data containers
SummarizedExperiment (SE) (Morgan et al. 2020) is a generic and highly optimized container for complex data structures. It has become a common choice for analyzing various types of biomedical profiling data, such as RNAseq, ChIp-Seq, microarrays, flow cytometry, proteomics, and single-cell sequencing.
[TreeSummarizedExperiment] (TreeSE) (Huang 2020) was developed as an extension to incorporate hierarchical information (such as phylogenetic trees and sample hierarchies) and reference sequences.
[MultiAssayExperiment] (MAE) (Ramos et al. 2017) provides an organized way to bind several different data containers together in a single object. For example, we can bind microbiome data (in TreeSE container) with metabolomic profiling data (in SE) container, with (partially) shared sample metadata. This is convenient and robust for instance, in subsetting and other data manipulation tasks. Microbiome data can be part of multiomics experiments and analysis strategies. We highlight how the methods used throughout in this book relate to this data framework by using the TreeSE, MAE, and classes beyond.
This section provides an introduction to these data containers. In microbiome data science, these containers link taxonomic abundance tables with rich side information on the features and samples. Taxonomic abundance data can be obtained by 16S rRNA amplicon or metagenomic sequencing, phylogenetic microarrays, or by other means. Many microbiome experiments include multiple versions and types of data generated independently or derived from each other through transformation or agglomeration. We start by providing recommendations on how to represent different varieties of multi-table data within the TreeSE class.
The options and recommendations are summarized in Table 2.1.
2.2.1 Assay data
The microbiome is the collection of all microbes (such as bacteria, viruses, fungi, etc.) in the body. When studying these microbes, data is needed, and that’s where assays come in.
An assay is a way of measuring the presence and abundance of different types of microbes in a sample. For example, if you want to know how many bacteria of a certain type are in your gut, you can use an assay to measure this. When storing assays, the original data is count-based. However, the original count-based taxonomic abundance tables may undergo different transformations, such as logarithmic, Centered Log-Ratio (CLR), or relative abundance. These are typically stored in assays.
Let us load example data and rename it as tse.
The assays slot contains the experimental data as multiple count matrices. The result of assays is a list of matrices.
assays(tse)
## List of length 1
## names(1): countsIndividual assays can be accessed via assay
assay(tse, "counts")[1:5,1:7]
## CL3 CC1 SV1 M31Fcsw M11Fcsw M31Plmr M11Plmr
## 549322 0 0 0 0 0 0 0
## 522457 0 0 0 0 0 0 0
## 951 0 0 0 0 0 0 1
## 244423 0 0 0 0 0 0 0
## 586076 0 0 0 0 0 0 0So, in summary, in the world of microbiome analysis, an assay is essentially a way to quantify and understand the composition of microbes in a given sample, which is super important for all kinds of research, ranging from human health to environment studies.
Furthermore, to illustrate the use of multiple assays, the relative abundance data can be calculated and stored along the original count data using transformAssay.
tse <- transformAssay(tse, assay.type = "counts", method = "relabundance")
assays(tse)
## List of length 2
## names(2): counts relabundanceNow there are two assays available in the tse object, counts and relabundance.
assay(tse, "relabundance")[1:5,1:7]
## CL3 CC1 SV1 M31Fcsw M11Fcsw M31Plmr M11Plmr
## 549322 0 0 0 0 0 0 0.000e+00
## 522457 0 0 0 0 0 0 0.000e+00
## 951 0 0 0 0 0 0 2.305e-06
## 244423 0 0 0 0 0 0 0.000e+00
## 586076 0 0 0 0 0 0 0.000e+00Here the dimension of the count data remains unchanged in transformation. This is in fact, a requirement for the assays.
2.2.2 colData
colData contains information about the samples used in the study. This information can include details such as the sample ID, the primers used in the analysis, the barcodes associated with the sample (truncated or complete), the type of sample (e.g. soil, fecal, mock) and a description of the sample.
colData(tse)
## DataFrame with 26 rows and 7 columns
## X.SampleID Primer Final_Barcode Barcode_truncated_plus_T
## <factor> <factor> <factor> <factor>
## CL3 CL3 ILBC_01 AACGCA TGCGTT
## CC1 CC1 ILBC_02 AACTCG CGAGTT
## SV1 SV1 ILBC_03 AACTGT ACAGTT
## M31Fcsw M31Fcsw ILBC_04 AAGAGA TCTCTT
## M11Fcsw M11Fcsw ILBC_05 AAGCTG CAGCTT
## ... ... ... ... ...
## TS28 TS28 ILBC_25 ACCAGA TCTGGT
## TS29 TS29 ILBC_26 ACCAGC GCTGGT
## Even1 Even1 ILBC_27 ACCGCA TGCGGT
## Even2 Even2 ILBC_28 ACCTCG CGAGGT
## Even3 Even3 ILBC_29 ACCTGT ACAGGT
## Barcode_full_length SampleType
## <factor> <factor>
## CL3 CTAGCGTGCGT Soil
## CC1 CATCGACGAGT Soil
## SV1 GTACGCACAGT Soil
## M31Fcsw TCGACATCTCT Feces
## M11Fcsw CGACTGCAGCT Feces
## ... ... ...
## TS28 GCATCGTCTGG Feces
## TS29 CTAGTCGCTGG Feces
## Even1 TGACTCTGCGG Mock
## Even2 TCTGATCGAGG Mock
## Even3 AGAGAGACAGG Mock
## Description
## <factor>
## CL3 Calhoun South Carolina Pine soil, pH 4.9
## CC1 Cedar Creek Minnesota, grassland, pH 6.1
## SV1 Sevilleta new Mexico, desert scrub, pH 8.3
## M31Fcsw M3, Day 1, fecal swab, whole body study
## M11Fcsw M1, Day 1, fecal swab, whole body study
## ... ...
## TS28 Twin #1
## TS29 Twin #2
## Even1 Even1
## Even2 Even2
## Even3 Even3To illustrate, X.SampleID gives the sample identifier, SampleType indicates the sample type (e.g. soil, fecal matter, control) and Description provides an additional description of the sample.
2.2.3 rowData
rowData contains data on the features of the analyzed samples. This is particularly important in the microbiome field for storing taxonomic information. This taxonomic information is extremely important for understanding the composition and diversity of the microbiome in each sample analyzed. It enables identification of the different types of microorganisms present in samples. It also allows you to explore the relationships between microbiome composition and various environmental or health factors.
rowData(tse)
## DataFrame with 19216 rows and 7 columns
## Kingdom Phylum Class Order Family
## <character> <character> <character> <character> <character>
## 549322 Archaea Crenarchaeota Thermoprotei NA NA
## 522457 Archaea Crenarchaeota Thermoprotei NA NA
## 951 Archaea Crenarchaeota Thermoprotei Sulfolobales Sulfolobaceae
## 244423 Archaea Crenarchaeota Sd-NA NA NA
## 586076 Archaea Crenarchaeota Sd-NA NA NA
## ... ... ... ... ... ...
## 278222 Bacteria SR1 NA NA NA
## 463590 Bacteria SR1 NA NA NA
## 535321 Bacteria SR1 NA NA NA
## 200359 Bacteria SR1 NA NA NA
## 271582 Bacteria SR1 NA NA NA
## Genus Species
## <character> <character>
## 549322 NA NA
## 522457 NA NA
## 951 Sulfolobus Sulfolobusacidocalda..
## 244423 NA NA
## 586076 NA NA
## ... ... ...
## 278222 NA NA
## 463590 NA NA
## 535321 NA NA
## 200359 NA NA
## 271582 NA NA2.2.4 rowTree
Phylogenetic trees also play an important role in the microbiome field. The TreeSE class can keep track of features and node relations via two functions, rowTree and rowLinks.
A tree can be accessed via rowTree as phylo object.
rowTree(tse)
##
## Phylogenetic tree with 19216 tips and 19215 internal nodes.
##
## Tip labels:
## 549322, 522457, 951, 244423, 586076, 246140, ...
## Node labels:
## , 0.858.4, 1.000.154, 0.764.3, 0.995.2, 1.000.2, ...
##
## Rooted; includes branch lengths.The links to the individual features are available through rowLinks.
rowLinks(tse)
## LinkDataFrame with 19216 rows and 5 columns
## nodeLab nodeNum nodeLab_alias isLeaf whichTree
## <character> <integer> <character> <logical> <character>
## 1 549322 1 alias_1 TRUE phylo
## 2 522457 2 alias_2 TRUE phylo
## 3 951 3 alias_3 TRUE phylo
## 4 244423 4 alias_4 TRUE phylo
## 5 586076 5 alias_5 TRUE phylo
## ... ... ... ... ... ...
## 19212 278222 19212 alias_19212 TRUE phylo
## 19213 463590 19213 alias_19213 TRUE phylo
## 19214 535321 19214 alias_19214 TRUE phylo
## 19215 200359 19215 alias_19215 TRUE phylo
## 19216 271582 19216 alias_19216 TRUE phyloPlease note that there can be a 1:1 relationship between tree nodes and features, but this is not a must-have. This means there can be features that are not linked to nodes, and nodes that are not linked to features. To change the links in an existing object, the changeTree function is available.
2.2.5 Alternative Experiments
Alternative experiments complement assays. They can contain complementary data, which is no longer tied to the same dimensions as the assay data. However, the number of samples (columns) must be the same.
This can come into play, for instance, when one has taxonomic abundance profiles quantified using different measurement technologies, such as phylogenetic microarrays, amplicon sequencing, or metagenomic sequencing. Another common use case is including abundance tables for different taxonomic ranks. Such alternative experiments concerning the same set of samples can be stored as
- Separate assays assuming that the taxonomic information can be mapped between features directly 1:1; or
- Data in the altExp slot of the
TreeSE, if the feature dimensions differ. Each element of the altExp slot is aSEor an object from a derived class with independent feature data.
The following shows how to store taxonomic abundance tables agglomerated at different taxonomic levels. However, the data could as well originate from entirely different measurement sources as long as the samples match.
Let us first agglomerate the data to Phylum level. This yields a new TreeSE data object.
tse_phylum <- agglomerateByRank(tse, "Phylum", na.rm=TRUE)
# Both have the same number of columns (samples)
dim(tse)
## [1] 19216 26
dim(tse_phylum)
## [1] 66 26Then we can add the new phylum-level data object as an alternative experiment in the original data.
# Add the new data object to the original data object as an alternative experiment with the name "Phylum"
altExp(tse, "Phylum") <- tse_phylum
# Check the alternative experiment names available in the data
altExpNames(tse)
## [1] "Phylum"We can now subset the data, for instance, and this acts on both altExp and assay data.
tse[,1:10]
## class: TreeSummarizedExperiment
## dim: 19216 10
## metadata(0):
## assays(2): counts relabundance
## rownames(19216): 549322 522457 ... 200359 271582
## rowData names(7): Kingdom Phylum ... Genus Species
## colnames(10): CL3 CC1 ... M31Tong M11Tong
## colData names(7): X.SampleID Primer ... SampleType Description
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(1): Phylum
## rowLinks: a LinkDataFrame (19216 rows)
## rowTree: 1 phylo tree(s) (19216 leaves)
## colLinks: NULL
## colTree: NULL
dim(altExp(tse[,1:10],"Phylum"))
## [1] 66 10For more details on altExp, you can check the introduction to the SingleCellExperiment package (Lun and Risso 2020).
2.2.6 MAEs
Multiple experiments relate to complementary measurement types, such as transcriptomic or metabolomic profiling of the microbiome or the host. Multiple experiments can be represented using the same options as alternative experiments, or by using the MAE class (Ramos et al. 2017). Depending on how the datasets relate to each other the data can be stored as:
- altExp if the samples can be matched directly 1:1; or
- As
MAEobjects, in which the connections between samples are defined through asampleMap. Each element on theexperimentsListof anMAEismatrixormatrix-like objects, includingSEobjects, and the number of samples can differ between the elements.
For information have a look at the intro vignette of the MultiAssayExperiment package.
| Option | Rows (features) | Cols (samples) | Recommended |
|---|---|---|---|
| assays | match | match | Data transformations |
| altExp | free | match | Alternative experiments |
| MultiAssay | free | free (mapping) | Multi-omic experiments |
2.3 Demonstration data
Open demonstration data for testing and benchmarking purposes is available from multiple locations. This chapter introduces some options. The other chapters of this book provide ample examples about the use of the data.
2.3.1 Package data
The mia R package contains example datasets that are direct conversions from the alternative phyloseq container to the TreeSE container.
List the available datasets in the mia package:
Load the GlobalPatterns data from the mia package:
data("GlobalPatterns", package="mia")
GlobalPatterns
## class: TreeSummarizedExperiment
## dim: 19216 26
## metadata(0):
## assays(1): counts
## rownames(19216): 549322 522457 ... 200359 271582
## rowData names(7): Kingdom Phylum ... Genus Species
## colnames(26): CL3 CC1 ... Even2 Even3
## colData names(7): X.SampleID Primer ... SampleType Description
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):
## rowLinks: a LinkDataFrame (19216 rows)
## rowTree: 1 phylo tree(s) (19216 leaves)
## colLinks: NULL
## colTree: NULLR packages contain additional demonstration data sets (see the Datasets section of the reference page):
2.3.2 ExperimentHub data
ExperimentHub provides a variety of data resources, including the microbiomeDataSets package (Morgan and Shepherd 2021; Lahti, Ernst, and Shetty 2021).
A table of the available datasets is available through the availableDataSets function.
library(microbiomeDataSets)
availableDataSets()
## Dataset
## 1 GrieneisenTSData
## 2 LahtiMLData
## 3 LahtiMData
## 4 OKeefeDSData
## 5 SilvermanAGutData
## 6 SongQAData
## 7 SprockettTHDataAll data are downloaded from ExperimentHub and cached for local re-use. Check the man pages of each function for a detailed documentation of the data contents and references. Let us retrieve a MAE dataset:
# mae <- HintikkaXOData()
# Since HintikkaXOData is now added to mia, we can load it directly from there
# We suggest to check other datasets from microbiomeDataSets
data(HintikkaXOData, package = "mia")
mae <- HintikkaXODataData is available in SE, r Biocpkg("TreeSE") and r Biocpkg("MAE") data containers; see the separate page on alternative containers for more details.
2.3.3 Curated metagenomic data
curatedMetagenomicData is a large collection of curated human microbiome datasets, provided as TreeSE objects (Pasolli et al. 2017). The resource provides curated human microbiome data including gene families, marker abundance, marker presence, pathway abundance, pathway coverage, and relative abundance for samples from different body sites. See the package homepage for more details on data availability and access.
As one example, let us retrieve the Vatanen (2016) (Vatanen et al. 2016) data set. This is a larger collection with a bit longer download time.
library(curatedMetagenomicData)
tse <- curatedMetagenomicData("Vatanen*", dryrun = FALSE, counts = TRUE)2.3.4 Human microbiome compendium
MicroBioMap dataset includes over 170k samples of publicly available 16S rRNA amplicon sequencing data, all processed using the same pipeline and reference database(Abdill2023?). After installing the MicroBioMap package (see the original website for instructions), you can load the compendium with
library(MicroBioMap)
cpd <- getCompendium()This returns a TreeSE object. Currently, the “tree” part of the TreeSE is not populated, but that is on the roadmap(compendiumpackage?).
After loading the compendium, you will have immediate access to nearly 170,000 microbiome samples of publicly available 16S rRNA amplicon sequencing data, all processed using the same pipeline and reference database. For more use examples in R/Bioconductor, see the MicroBioMap vignette.
2.3.5 Other data sources
The current collections provide access to vast microbiome data resources. The output has to be converted into TreeSE/MAE separately.
2.4 Loading experimental microbiome data
2.4.1 16S workflow
Result of amplicon sequencing is a large number of files that include all the sequences that were read from samples. Those sequences need to be matched with taxa. Additionally, we need to know how many times each taxa were found from each sample.
There are several algorithms to do that, and DADA2 is one of the most common. You can find DADA2 pipeline tutorial, for example, here. After the DADA2 portion of the tutorial is completed, the data is stored into phyloseq object (Bonus: Handoff to phyloseq). To store the data to TreeSE, follow the example below.
You can find full workflow script without further explanations and comments from here
Load required packages.
Create arbitrary example sample metadata like it was done in the tutorial. Usually, sample metadata is imported as a file.
samples.out <- rownames(seqtab.nochim)
subject <- sapply(strsplit(samples.out, "D"), `[`, 1)
gender <- substr(subject,1,1)
subject <- substr(subject,2,999)
day <- as.integer(sapply(strsplit(samples.out, "D"), `[`, 2))
samdf <- data.frame(Subject=subject, Gender=gender, Day=day)
samdf$When <- "Early"
samdf$When[samdf$Day>100] <- "Late"
rownames(samdf) <- samples.outConvert data into right format and create a TreeSE object.
# Create a list that contains assays
counts <- t(seqtab.nochim)
counts <- as.matrix(counts)
assays <- SimpleList(counts = counts)
# Convert colData and rowData into DataFrame
samdf <- DataFrame(samdf)
taxa <- DataFrame(taxa)
# Create TreeSE
tse <- TreeSummarizedExperiment(assays = assays,
colData = samdf,
rowData = taxa
)
# Remove mock sample like it is also done in DADA2 pipeline tutorial
tse <- tse[ , colnames(tse) != "mock"]Add sequences into referenceSeq slot and convert rownames into simpler format.
# Convert sequences into right format
dna <- Biostrings::DNAStringSet( rownames(tse) )
# Add sequences into referenceSeq slot
referenceSeq(tse) <- dna
# Convert rownames into ASV_number format
rownames(tse) <- paste0("ASV", seq( nrow(tse) ))
tse
## class: TreeSummarizedExperiment
## dim: 232 20
## metadata(0):
## assays(1): counts
## rownames(232): ASV1 ASV2 ... ASV231 ASV232
## rowData names(7): Kingdom Phylum ... Genus Species
## colnames(20): F3D0 F3D1 ... F3D9 Mock
## colData names(4): Subject Gender Day When
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):
## rowLinks: NULL
## rowTree: NULL
## colLinks: NULL
## colTree: NULL
## referenceSeq: a DNAStringSet (232 sequences)2.4.2 Import from external files
Microbiome (taxonomic) profiling data is commonly distributed in various file formats. You can import such external data files as a TreeSE object, but the details depend on the file format. Here, we provide examples for common formats. Some datasets and raw files to learn how to import raw data and construct TreeSE/MAE containers are available in the microbiome data repository.
2.4.2.1 CSV import
CSV data tables can be imported with the standard R functions, then converted to the desired format. For detailed examples, you can check the Bioconductor course material by Martin Morgan. You can also check the example files and construct your own CSV files accordingly.
Recommendations for the CSV files are the following. File names are arbitrary; we refer here to the same names as in the examples:
Abundance table (
assay_taxa.csv): data matrix (features x samples); first column provides feature IDs, the first row provides sample IDs; other values should be numeric (abundances).Row data (
rowdata_taxa.csv): data table (features x info); first column provides feature IDs, the first row provides column headers; this file usually contains the taxonomic mapping between different taxonomic levels. Ideally, the feature IDs (row names) match one-to-one with the abundance table row names.Column data (
coldata.csv): data table (samples x info); first column provides sample IDs, the first row provides column headers; this file usually contains the sample metadata/phenodata (such as subject age, health etc). Ideally, the sample IDs match one-to-one with the abundance table column names.
After you have set up the CSV files, you can read them in R:
count_file <- system.file("extdata", "assay_taxa.csv", package = "OMA")
tax_file <- system.file("extdata", "rowdata_taxa.csv", package = "OMA")
sample_file <- system.file("extdata", "coldata.csv", package = "OMA")
# Load files
counts <- read.csv(count_file, row.names=1) # Abundance table (e.g. ASV data; to assay data)
tax <- read.csv(tax_file, row.names=1) # Taxonomy table (to rowData)
samples <- read.csv(sample_file, row.names=1) # Sample data (to colData)After reading the data in R, ensure the following:
abundance table (
counts): numericmatrix, with feature IDs as rownames and sample IDs as column names.rowdata (
tax):DataFrame, with feature IDs as rownames. If this is adata.frameyou can use the functionDataFrame()to change the format. Column names are free but in microbiome analysis they usually they refer to taxonomic ranks. The rownames in rowdata should match with rownames in abundance table.coldata (
samples):DataFrame, with sample IDs as rownames. If this is adata.frameyou can use the functionDataFrame()to change the format. Column names are free. The rownames incoldatashould match with colnames in abundance table.
Always ensure that the tables have rownames! The TreeSE constructor compares rownames and ensures that, for example, right samples are linked with right patient.
Also ensure that the row and column names match one-to-one between abundance table, rowdata, and coldata:
If you hesitate about the format of the data, you can compare to one of the available demonstration datasets, and make sure that your data components have the same format.
There are many different source files and many different ways to read data in R. One can do data manipulation in R as well. Investigate the entries as follows.
# coldata rownames match assay colnames
all(rownames(samples) == colnames(counts)) # our dataset
## [1] TRUE
class(samples) # should be data.frame or DataFrame
## [1] "data.frame"
# rowdata rownames match assay rownames
all(rownames(tax) == rownames(counts)) # our dataset
## [1] TRUE
class(tax) # should be data.frame or DataFrame
## [1] "data.frame"
# Counts
class(counts) # should be a numeric matrix
## [1] "matrix" "array"2.4.3 Constructing TreeSE
Now, let’s create the TreeSE object from the input data tables. Here we also convert the data objects in their preferred formats:
- counts –> numeric matrix
- rowData –> DataFrame
- colData –> DataFrame
The SimpleList could be used to include multiple alternative assays, if necessary.
# Create a TreeSE
tse_taxa <- TreeSummarizedExperiment(assays = SimpleList(counts = counts),
colData = DataFrame(samples),
rowData = DataFrame(tax))
tse_taxa
## class: TreeSummarizedExperiment
## dim: 12706 40
## metadata(0):
## assays(1): counts
## rownames(12706): GAYR01026362.62.2014 CVJT01000011.50.2173 ...
## JRJTB:03787:02429 JRJTB:03787:02478
## rowData names(7): Phylum Class ... Species OTU
## colnames(40): C1 C2 ... C39 C40
## colData names(6): Sample Rat ... Fat XOS
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):
## rowLinks: NULL
## rowTree: NULL
## colLinks: NULL
## colTree: NULLNow you should have a ready-made TreeSE data object that can be used in downstream analyses.
2.4.4 Constructing MAE
To construct a MAE object, just combine multiple TreeSE data containers. Here we import metabolite data from the same study.
count_file <- system.file("extdata", "assay_metabolites.csv", package = "OMA")
sample_file <- system.file("extdata", "coldata.csv", package = "OMA")
# Load files
counts <- read.csv(count_file, row.names=1)
samples <- read.csv(sample_file, row.names=1)
# Create a TreeSE for the metabolite data
tse_metabolite <- TreeSummarizedExperiment(assays = SimpleList(concs = as.matrix(counts)),
colData = DataFrame(samples))
tse_metabolite
## class: TreeSummarizedExperiment
## dim: 38 40
## metadata(0):
## assays(1): concs
## rownames(38): Butyrate Acetate ... Malonate 1,3-dihydroxyacetone
## rowData names(0):
## colnames(40): C1 C2 ... C39 C40
## colData names(6): Sample Rat ... Fat XOS
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):
## rowLinks: NULL
## rowTree: NULL
## colLinks: NULL
## colTree: NULLNow we can combine these two experiments into MAE.
# Create an ExperimentList that includes experiments
experiments <- ExperimentList(microbiome = tse_taxa,
metabolite = tse_metabolite)
# Create a MAE
mae <- MultiAssayExperiment(experiments = experiments)
mae
## A MultiAssayExperiment object of 2 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 2:
## [1] microbiome: TreeSummarizedExperiment with 12706 rows and 40 columns
## [2] metabolite: TreeSummarizedExperiment with 38 rows and 40 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat files2.4.5 Import functions for standard formats
Specific import functions are provided for:
- Biom files (see
help(mia::importBIOM)) - QIIME2 files (see
help(mia::importQIIME2)) - Mothur files (see
help(mia::importMothur))
2.4.5.1 Biom import
Here we show how Biom files are imported into a TreeSE object using as an example Tengeler2020, which is further described here. This dataset consists of 3 files, which can be fetched or downloaded from this repository:
- biom file: abundance table and taxonomy information
- csv file: sample metadata
- tree file: phylogenetic tree
To begin with, we store the data in a local directory within the working directory, such as data/, and define the source file paths.
biom_file_path <- system.file("extdata", "Aggregated_humanization2.biom", package = "OMA")
sample_meta_file_path <- system.file("extdata", "Mapping_file_ADHD_aggregated.csv", package = "OMA")
tree_file_path <- system.file("extdata", "Data_humanization_phylo_aggregation.tre", package = "OMA")Now we can read in the biom file and convert it into a TreeSE object. In addition, we retrieve the rank names from the prefixes of the feature names and then remove them with the rank.from.prefix and prefix.rm optional arguments.
library(mia)
# read biom and convert it to TreeSE
tse <- importBIOM(biom_file_path,
rank.from.prefix = TRUE,
prefix.rm = TRUE)
# Check
tse
## class: TreeSummarizedExperiment
## dim: 151 27
## metadata(0):
## assays(1): counts
## rownames(151): 1726470 1726471 ... 17264756 17264757
## rowData names(6): taxonomy1 phylum ... family genus
## colnames(27): A110 A111 ... A38 A39
## colData names(0):
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):
## rowLinks: NULL
## rowTree: NULL
## colLinks: NULL
## colTree: NULLThe assays slot includes a list of abundance tables. The imported abundance table is named as “counts”. Let us inspect only the first cols and rows.
assay(tse, "counts")[1:3, 1:3]
## A110 A111 A12
## 1726470 17722 11630 0
## 1726471 12052 0 2679
## 17264731 0 970 0The rowdata includes taxonomic information from the biom file. The head() command shows just the beginning of the data table for an overview.
knitr::kable() helps print the information more nicely.
head(rowData(tse))
## DataFrame with 6 rows and 6 columns
## taxonomy1 phylum class order
## <character> <character> <character> <character>
## 1726470 "k__Bacteria Bacteroidetes Bacteroidia Bacteroidales
## 1726471 "k__Bacteria Bacteroidetes Bacteroidia Bacteroidales
## 17264731 "k__Bacteria Bacteroidetes Bacteroidia Bacteroidales
## 17264726 "k__Bacteria Bacteroidetes Bacteroidia Bacteroidales
## 1726472 "k__Bacteria Verrucomicrobia Verrucomicrobiae Verrucomicrobiales
## 17264724 "k__Bacteria Bacteroidetes Bacteroidia Bacteroidales
## family genus
## <character> <character>
## 1726470 Bacteroidaceae Bacteroides"
## 1726471 Bacteroidaceae Bacteroides"
## 17264731 Porphyromonadaceae Parabacteroides"
## 17264726 Bacteroidaceae Bacteroides"
## 1726472 Verrucomicrobiaceae Akkermansia"
## 17264724 Bacteroidaceae Bacteroides"We further polish the feature names by removing unnecessary characters and then replace the original rowData with its updated version.
# Genus level has additional '\"', so let's delete that also
rowdata_modified <- BiocParallel::bplapply(rowData(tse),
FUN = stringr::str_remove,
pattern = '\"')
# rowdata_modified is a list, so convert this back to DataFrame format.
# and assign the cleaned data back to the TSE rowData
rowData(tse) <- DataFrame(rowdata_modified)
# Now we have a nicer table
head(rowData(tse))
## DataFrame with 6 rows and 6 columns
## taxonomy1 phylum class order
## <character> <character> <character> <character>
## 1726470 k__Bacteria Bacteroidetes Bacteroidia Bacteroidales
## 1726471 k__Bacteria Bacteroidetes Bacteroidia Bacteroidales
## 17264731 k__Bacteria Bacteroidetes Bacteroidia Bacteroidales
## 17264726 k__Bacteria Bacteroidetes Bacteroidia Bacteroidales
## 1726472 k__Bacteria Verrucomicrobia Verrucomicrobiae Verrucomicrobiales
## 17264724 k__Bacteria Bacteroidetes Bacteroidia Bacteroidales
## family genus
## <character> <character>
## 1726470 Bacteroidaceae Bacteroides
## 1726471 Bacteroidaceae Bacteroides
## 17264731 Porphyromonadaceae Parabacteroides
## 17264726 Bacteroidaceae Bacteroides
## 1726472 Verrucomicrobiaceae Akkermansia
## 17264724 Bacteroidaceae BacteroidesWe notice that the imported biom file did not contain any colData yet, so only an empty dataframe appears in this slot.
Let us add colData from the sample metadata, which is stored in a CSV file.
# CSV file with colnames in the first row and rownames in the first column
sample_meta <- read.csv(sample_meta_file_path,
sep = ",", row.names = 1)
# Add this sample data to colData of the taxonomic data object
# Note that the data must be given in a DataFrame format (required for our purposes)
colData(tse) <- DataFrame(sample_meta)Now the colData includes the sample metadata.
head(colData(tse))
## DataFrame with 6 rows and 4 columns
## Treatment Cohort TreatmentxCohort Description
## <character> <character> <character> <character>
## A110 ADHD Cohort_1 ADHD_Cohort_1 A110
## A12 ADHD Cohort_1 ADHD_Cohort_1 A12
## A15 ADHD Cohort_1 ADHD_Cohort_1 A15
## A19 ADHD Cohort_1 ADHD_Cohort_1 A19
## A21 ADHD Cohort_2 ADHD_Cohort_2 A21
## A23 ADHD Cohort_2 ADHD_Cohort_2 A23Finally, we add a phylogenetic tree to the rowData slot. Such feature is available only in TreeSE objects. Similarly, Trees specifying the sample hierarchy can be stored in the colTree slot.
Here, we read in the file containing the phylogenetic tree and insert it in corresponding slot of the TreeSE object.
# Reads the tree file
tree <- ape::read.tree(tree_file_path)
# Add tree to rowTree
rowTree(tse) <- tree
# Check
tse
## class: TreeSummarizedExperiment
## dim: 151 27
## metadata(0):
## assays(1): counts
## rownames(151): 1726470 1726471 ... 17264756 17264757
## rowData names(6): taxonomy1 phylum ... family genus
## colnames(27): A110 A12 ... A35 A38
## colData names(4): Treatment Cohort TreatmentxCohort Description
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):
## rowLinks: a LinkDataFrame (151 rows)
## rowTree: 1 phylo tree(s) (151 leaves)
## colLinks: NULL
## colTree: NULLNow the rowTree slot contains the phylogenetic tree:
2.4.6 Conversions between data formats in R
If the data has already been imported in R in another format, it can be readily converted into TreeSE, as shown in our next example. Note that similar conversion functions to TreeSE are available for multiple data formats via the mia package (see convertFrom* for phyloseq, Biom, and DADA2).
library(mia)
# phyloseq example data
data(GlobalPatterns, package="phyloseq")
GlobalPatterns_phyloseq <- GlobalPatterns
GlobalPatterns_phyloseq
## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 19216 taxa and 26 samples ]
## sample_data() Sample Data: [ 26 samples by 7 sample variables ]
## tax_table() Taxonomy Table: [ 19216 taxa by 7 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 19216 tips and 19215 internal nodes ]# convert phyloseq to TSE
GlobalPatterns_TSE <- convertFromPhyloseq(GlobalPatterns_phyloseq)
GlobalPatterns_TSE
## class: TreeSummarizedExperiment
## dim: 19216 26
## metadata(0):
## assays(1): counts
## rownames(19216): 549322 522457 ... 200359 271582
## rowData names(7): Kingdom Phylum ... Genus Species
## colnames(26): CL3 CC1 ... Even2 Even3
## colData names(7): X.SampleID Primer ... SampleType Description
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):
## rowLinks: a LinkDataFrame (19216 rows)
## rowTree: 1 phylo tree(s) (19216 leaves)
## colLinks: NULL
## colTree: NULLWe can also convert TreeSE objects into phyloseq with respect to the shared components that are supported by both formats (i.e. taxonomic abundance table, sample metadata, taxonomic table, phylogenetic tree, sequence information). This is useful for instance when additional methods are available for phyloseq.
# convert TSE to phyloseq
GlobalPatterns_phyloseq2 <- convertToPhyloseq(GlobalPatterns_TSE)
GlobalPatterns_phyloseq2
## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 19216 taxa and 26 samples ]
## sample_data() Sample Data: [ 26 samples by 7 sample variables ]
## tax_table() Taxonomy Table: [ 19216 taxa by 7 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 19216 tips and 19215 internal nodes ]Conversion is possible between other data formats. Interested readers can refer to the following functions:
2.5 Exporting data container
2.5.1 Transforming a TreeSummarizedExperiment into a dataframe
Transforming a TreeSE object into a dataframe is straightforward with the mia package. The meltSE function is particularly handy for this purpose. It allows you to melt various parts of a TreeSE object into a dataframe based on the parameters you specify.
2.5.2 Export TreeSummarizedExperiment
Exporting a TreeSE data container can be done using feather package. TreeSE object has to be converted into a dataframe (data.frame and not DataFrame) . The output file is a .feather file, which can be imported in other languages such as Julia or Python. For information, have a look at the FeatherFile Julia package and feather-format Python library.
Another way could be using a CSV file. This works the same as for a feather file, make sure you have converted your TreeSE data container as a dataframe. Here note that you can decide whether you want to write the row names or not.
write.csv(molten_tse, "path/to/tse.csv", row.names=FALSE)2.5.3 Export MultiAssayExperiment
Exporting a MultiAssayExperiment data container can also be done using feather package.
# Convert into a data.frame
molten_mae <- longFormat(mae)
molten_mae <- data.frame(molten_mae)
path <- "path/to/mae.feather"
write_feather(molten_mae,path)And as a CSV file.
write.csv(molten_mae, "path/to/mae.csv", row.names=FALSE)