3 Data containers
This section provides an introduction to the TreeSummarizedExperiment (TreeSE) and MultiAssayExperiment (MAE) data containers introduced in Section 1.2. In microbiome data science, these containers link taxonomic abundance tables with rich side information on the features and samples.
One key advantage of the SummarizedExperiment ecosystem is its flexibility and generalizability. While taxonomic features remain central in microbiome studies, there is growing interest in analyzing and integrating diverse types of features. These may include:
Taxonomic features (also referred to as taxa, microbes, or taxonomic profile), typically obtained through 16S rRNA amplicon sequencing, shotgun metagenomic sequencing, or phylogenetic microarrays. These represent the composition and abundance of microbial taxa within a sample.
Functional features (also known as gene abundance, pathway abundance, or functional profile), usually derived from shotgun metagenomic sequencing. These describe the functional potential of microbial communities within a sample.
Transcriptomic features (or metatranscriptomic profile), typically obtained using metatranscriptomic sequencing (RNA-seq). These capture the actively expressed genes in microbial communities and reflect functional activity within a sample.
Metabolomic features, typically obtained using mass spectrometry (MS) or nuclear magnetic resonance (NMR). These represent small molecules produced or consumed by the microbiome within a sample.
Proteomic features, typically obtained using mass spectrometry (MS). These capture the expressed proteins in a sample and reflect microbial activity within a sample.
In addition, these data tables often exist in multiple versions, derived through transformations or agglomerations. We start by providing recommendations on how to represent different varieties of multi-table data within the TreeSE class. The options and recommendations are summarized in Table 3.1.
3.1 Structure of TreeSE
TreeSE contains several distinct slots, each holding a specific type of data. The assays slot is the core of TreeSE, storing abundance tables that contain the counts or concentrations of features in each sample. Features can be taxonomic, metabolomic, antimicrobial resistance genes, or other measured entities, and are represented as rows. The columns correspond to unique samples.
Building upon the assays, TreeSE accommodates various data types for both features and samples. In rowData, the rows correspond to the same features (rows) as in the abundance tables, while the columns represent variables such as taxonomy ranks. Similarly, in colData, each row matches the samples (columns) from the abundance tables, with the columns of colData containing metadata like disease status or patient ID and time point if the dataset includes time series.
The slots in TreeSE are outlined below:
-
assays: Stores a list of abundance tables. Each table has consistent rows and columns, where rows represent features and columns represent samples. -
rowData: Contains metadata about the rows (features). For example, this slot can include a taxonomy table. -
colData: Holds metadata about the columns (samples), such as patient information or the time points when samples were collected. -
rowTree: Stores a hierarchical tree for the rows, such as a phylogenetic tree representing the relationships between features. -
colTree: Includes a hierarchical tree for the columns, which can represent relationships between samples, for example, indicating whether patients are relatives and the structure of those relationships. -
rowLinks: Contains information about the linkages between rows and the nodes in therowTree. -
colLinks: Contains information about the linkages between columns and the nodes in thecolTree. -
referenceSeq: Holds reference sequences, i.e., the sequences that correspond to each taxon identified in the rows. -
metadata: Contains metadata about the experiment, such as the date it was conducted and the researchers involved.
These slots are illustrated in the figure below:
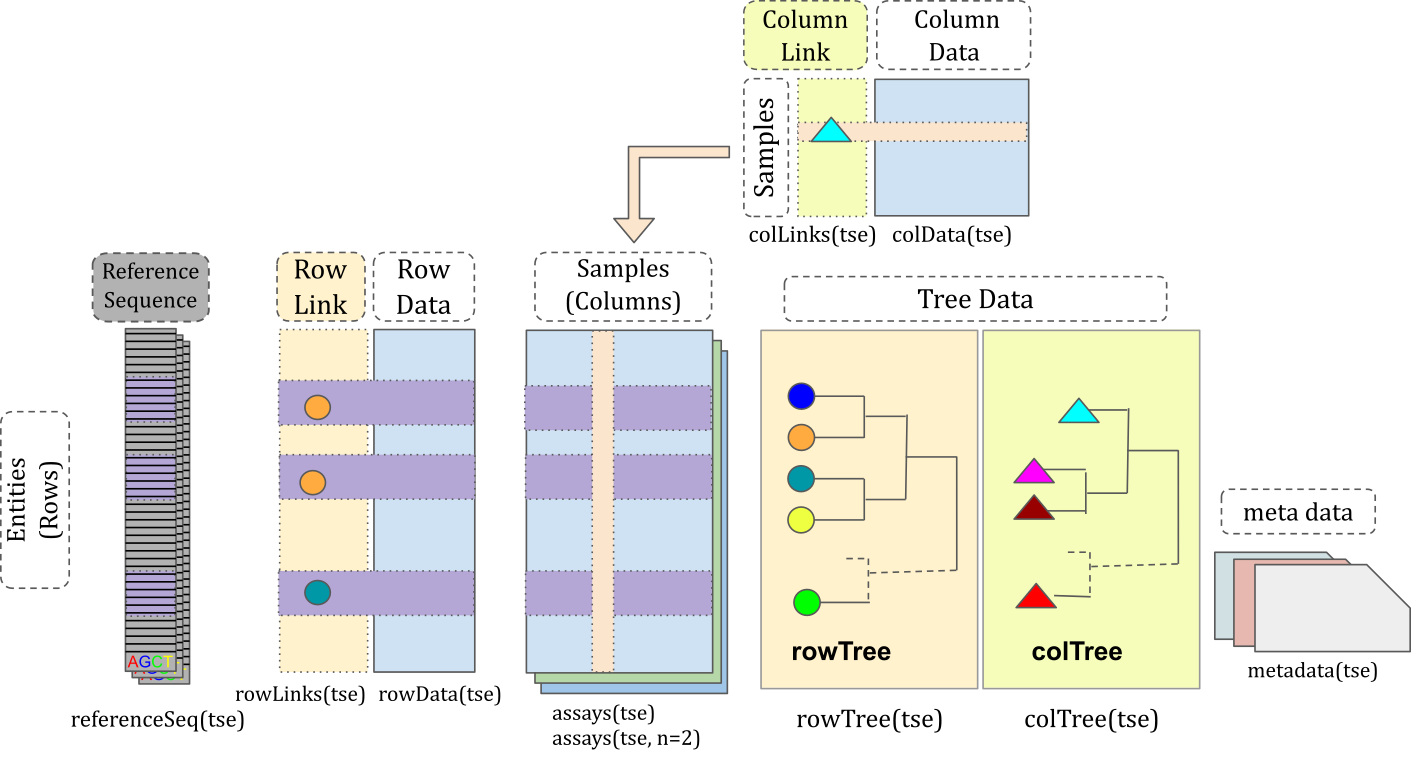
Additionally, TreeSE includes:
-
reducedDim: Contains reduced dimensionality representations of the samples, such as Principal Component Analysis (PCA) results (see Chapter 14). -
altExp: Stores alternative experiments, which areTreeSEobjects sharing the same samples but with different feature sets.
Among these, assays, rowData, colData, and metadata are shared with the SummarizedExperiment (SE) data container. reducedDim and altExp come from inheriting the SingleCellExperiment (SCE) class. The rowTree, colTree, rowLinks, colLinks, and referenceSeq slots are unique to TreeSE.
3.2 Rows and columns
Let us load example data and store it in variable tse.
library(mia)
data("GlobalPatterns", package = "mia")
tse <- GlobalPatterns
tse
## class: TreeSummarizedExperiment
## dim: 19216 26
## metadata(0):
## assays(1): counts
## rownames(19216): 549322 522457 ... 200359 271582
## rowData names(7): Kingdom Phylum ... Genus Species
## colnames(26): CL3 CC1 ... Even2 Even3
## colData names(7): X.SampleID Primer ... SampleType Description
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):
## rowLinks: a LinkDataFrame (19216 rows)
## rowTree: 1 phylo tree(s) (19216 leaves)
## colLinks: NULL
## colTree: NULLThe TreeSE object, similar to a standard data.frame or matrix, has rows and columns. Typically, samples are stored in columns, while features are stored in rows. You can extract subsets of the data, such as the first five rows (first five features) and certain three columns (certain three samples). The object manages the linkages between data (e.g., the assay data and the sample metadata), ensuring that when you subset the data object, all its parts are subsetted simultaneously, such that they remain matched with each other.
tse <- tse[1:5, c(1, 19, 16)]
tse
## class: TreeSummarizedExperiment
## dim: 5 3
## metadata(0):
## assays(1): counts
## rownames(5): 549322 522457 951 244423 586076
## rowData names(7): Kingdom Phylum ... Genus Species
## colnames(3): CL3 TRRsed1 NP2
## colData names(7): X.SampleID Primer ... SampleType Description
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):
## rowLinks: a LinkDataFrame (5 rows)
## rowTree: 1 phylo tree(s) (19216 leaves)
## colLinks: NULL
## colTree: NULLCompared to the original data the dimensions are for rows and columns 5 and 3, respectively.
SummarizedExperiment objects have rows and columns. Also MultiAssayExperiment, introduced in Section 3.8, have rows and columns but the structure is more complicated. You can find more examples on subsetting from Chapter 9.
3.3 Assay data
When studying microbiomes, the primary type of data is in the form of abundance of given microbes in given samples. This sample per feature table forms the core of the TreeSE object, called the ‘assay data’.
An assay is a measurement of the presence and abundance of different taxonomic features in a sample. The assay data records this in a table where rows are unique features and columns are unique samples and each entry contain a number describing the amount a given feature is present in a given sample. Note that when storing assays, the original data is count-based. However, due to the nature of how microbiome data is produced, these count-based abundances rarely reflect the true counts of features in the sample, and thus the abundance tables often undergo different transformations, such as logarithmic, Centered Log-Ratio (CLR), or relative abundance to make these abundance values comparable with each other. See Chapter 11 for more information on transformations.
The microbial abundance tables are stored in assays. The assays slot contains the abundance data as multiple count matrices. The result of assays is a list of matrices.
assays(tse)
## List of length 1
## names(1): countsIndividual assays can be accessed via assay.
In summary, in the world of microbiome analysis, an assay is essentially a way to describe the composition of microbes in a given sample. This way we can summarize the microbiome profile of a human gut or a sample of soil.
Furthermore, to illustrate the use of multiple assays, we can create an empty matrix and add it to the object.
Now there are two assays available in the tse object, counts and empty_table.
Here the dimension of the assay data remains unchanged. This is, in fact, a requirement for the assays.
3.4 colData
colData contains information about the samples used in the study. This sample metadata can include details such as the sample ID, the primers used in the analysis, the barcodes associated with the sample (truncated or complete), the type of sample (e.g. soil, fecal, mock) and a description of the sample.
colData(tse)
## DataFrame with 3 rows and 7 columns
## X.SampleID Primer Final_Barcode Barcode_truncated_plus_T
## <factor> <factor> <factor> <factor>
## CL3 CL3 ILBC_01 AACGCA TGCGTT
## TRRsed1 TRRsed1 ILBC_22 ACATGT ACATGT
## NP2 NP2 ILBC_19 ACAGTT AACTGT
## Barcode_full_length SampleType
## <factor> <factor>
## CL3 CTAGCGTGCGT Soil
## TRRsed1 CACGTGACATG Sediment (estuary)
## NP2 TCGCGCAACTG Ocean
## Description
## <factor>
## CL3 Calhoun South Carolina Pine soil, pH 4.9
## TRRsed1 Tijuana River Reserve, depth 1
## NP2 Newport Pier, CA surface water, Time 1To illustrate, X.SampleID gives the sample identifier, SampleType indicates the sample type (e.g. soil, fecal matter, control) and Description provides an additional description of the sample.
3.5 rowData
rowData contains data on the features, such as taxonomic features of the analyzed samples. This is particularly important in the microbiome field for storing taxonomic information, such as the Species, Genus or Family of the different microorganisms present in samples. This taxonomic information is extremely important for understanding the composition and diversity of the microbiome in each sample.
rowData(tse)
## DataFrame with 5 rows and 7 columns
## Kingdom Phylum Class Order Family
## <character> <character> <character> <character> <character>
## 549322 Archaea Crenarchaeota Thermoprotei NA NA
## 522457 Archaea Crenarchaeota Thermoprotei NA NA
## 951 Archaea Crenarchaeota Thermoprotei Sulfolobales Sulfolobaceae
## 244423 Archaea Crenarchaeota Sd-NA NA NA
## 586076 Archaea Crenarchaeota Sd-NA NA NA
## Genus Species
## <character> <character>
## 549322 NA NA
## 522457 NA NA
## 951 Sulfolobus Sulfolobusacidocalda..
## 244423 NA NA
## 586076 NA NA3.6 rowTree
Phylogenetic trees play an important role in the microbiome field. Many times it is useful to know how closely related the taxonomic features present in the data are. For example, to calculate widely-used phylogenetically weighted microbiome dissimilarity metrics such as UniFrac, we need information on not only the presence and abundance of taxonomic features in each sample but also the evolutionary relatedness among these taxonomic features. The TreeSE class can keep track of relations among features via two functions, rowTree and rowLinks.
A tree can be accessed via rowTree as phylo object.
rowTree(tse)
##
## Phylogenetic tree with 19216 tips and 19215 internal nodes.
##
## Tip labels:
## 549322, 522457, 951, 244423, 586076, 246140, ...
## Node labels:
## , 0.858.4, 1.000.154, 0.764.3, 0.995.2, 1.000.2, ...
##
## Rooted; includes branch length(s).Each row in TreeSE is linked to a specific node in a tree. This relationship is stored in the rowLinks slot, which has the same rows as TreeSE. The rowLinks slot contains information about which tree node corresponds to each row and whether the node is a leaf (tip) or an internal node, among other details.
rowLinks(tse)
## LinkDataFrame with 5 rows and 5 columns
## nodeLab nodeNum nodeLab_alias isLeaf whichTree
## <character> <integer> <character> <logical> <character>
## 1 549322 1 alias_1 TRUE phylo
## 2 522457 2 alias_2 TRUE phylo
## 3 951 3 alias_3 TRUE phylo
## 4 244423 4 alias_4 TRUE phylo
## 5 586076 5 alias_5 TRUE phyloPlease note that there can be a 1:1 relationship between tree nodes and features; however, this is not required. This means there can be features that are not linked to nodes, and nodes that are not linked to features. To change the links in an existing object, the changeTree() function is available.
3.7 Alternative experiments
Alternative experiments (altExp) complement assays. They can contain complementary data, which is no longer tied to the same dimensions as the assay data. However, the number of samples (columns) must be the same.
This can come into play, for instance, when one has taxonomic abundance profiles quantified using different measurement technologies, such as phylogenetic microarrays, amplicon sequencing, or metagenomic sequencing. Another common use case is including abundance tables for different taxonomic ranks. Such alternative experiments concerning the same set of samples can be stored as
- Separate assays assuming that the taxonomic information can be mapped between features directly 1:1; or
- Data in the
altExpslot of theTreeSE, if the feature dimensions differ. Each element of thealtExpslot is aSEor an object from a derived class with independent feature data.
The following shows how to store taxonomic abundance tables agglomerated at different taxonomic levels. However, the data could as well originate from entirely different measurement sources (e.g., 16S amplicon and metagenomic sequence data) as long as the samples match.
Let us first subset the data so that it has only two rows.
Then we can add the new data object as an alternative experiment in the original data.
# Add the new data object to the original data object as an alternative
# experiment with the specified name
altExp(tse, "subsetted") <- tse_sub
# Retrieve and display the names of alternative experiments available
altExpNames(tse)
## [1] "subsetted"Now, if we subset the data, this acts on both the altExp and the assay data.
For more details on altExp, you can check the introduction to the SingleCellExperiment package (Lun and Risso 2020).
3.8 Multiple experiments
Multiple experiments relate to complementary measurement types from the same samples, such as transcriptomic or metabolomic profiling of the microbiome. Multiple experiments can be represented using the same options as alternative experiments, or by using the MAE class (Ramos et al. 2017). Depending on how the datasets relate to each other the data can be stored as:
-
altExpif the samples can be matched directly 1:1; or - As
MAEobjects, in which the connections between samples are defined through asampleMap. Each element on theExperimentListof anMAEismatrixormatrix-like objects, includingSEobjects, and the number of samples can differ between the elements.
In a MAE, the “subjects” represent patients. The MAE has four main slots, with experiments being the core. This slot holds a list of experiments, each in (Tree)SE format. To handle complex mappings between samples (observations) across different experiments, the sampleMap slot stores information about how each sample in the experiments is linked to a patient. Metadata for each patient is stored in the colData slot. Unlike the colData in TreeSE, this colData is meant to store only metadata that remains constant throughout the trial.
-
experiments: Contains experiments, such as different omics data, inTreeSEformat. -
sampleMap: Holds linkages between patients (subjects) and samples in the experiments (observations). -
colData: Includes patient metadata that remains unchanged throughout the trial.
These slots are illustrated in the figure below:
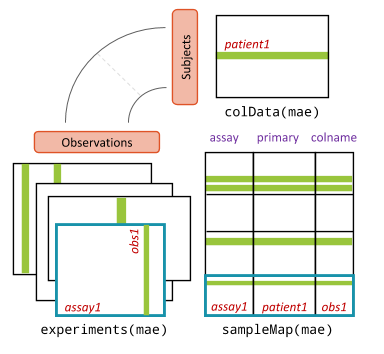
Additionally, the object includes a metadata slot that contains information about the dataset, such as the trial period and the creator of the MAE object.
The MAE object can handle more complex relationships between experiments. It manages the linkages between samples and experiments, ensuring that the data remains consistent and well-organized.
data("HintikkaXOData")
mae <- HintikkaXOData
mae
## A MultiAssayExperiment object of 3 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 3:
## [1] microbiota: TreeSummarizedExperiment with 12706 rows and 40 columns
## [2] metabolites: TreeSummarizedExperiment with 38 rows and 40 columns
## [3] biomarkers: TreeSummarizedExperiment with 39 rows and 40 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesThe sampleMap is a crucial component of the MAE object as it acts as an important bookkeeper, maintaining the information about which samples are associated with which experiments. This ensures that data linkages are correctly managed and preserved across different types of measurements.
For illustration, let’s subset the data by taking first five samples.
mae <- mae[, 1:5, ]
mae
## A MultiAssayExperiment object of 3 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 3:
## [1] microbiota: TreeSummarizedExperiment with 12706 rows and 5 columns
## [2] metabolites: TreeSummarizedExperiment with 38 rows and 5 columns
## [3] biomarkers: TreeSummarizedExperiment with 39 rows and 5 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesIf you have multiple experiments containing multiple measures from same sources (e.g., patients/host, individuals/sites), you can utilize the MultiAssayExperiment object to keep track of which samples belong to which patient.
The following dataset illustrates how to utilize the sample mapping system in MAE. It includes two omics data: biogenic amines and fatty acids, collected from 10 chickens.
mae <- readRDS(system.file("extdata", "mae_holofood.Rds", package = "OMA"))
mae
## A MultiAssayExperiment object of 2 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 2:
## [1] BIOGENIC AMINES: TreeSummarizedExperiment with 7 rows and 14 columns
## [2] FATTY ACIDS: TreeSummarizedExperiment with 19 rows and 19 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesWe can see that there are more than ten samples per omic dataset due to multiple samples from different time points collected for some animals. From the colData of MAE, we can observe the individual animal metadata, including information that remains constant throughout the trial.
colData(mae)
## DataFrame with 10 rows and 13 columns
## marker.canonical_url marker.type Treatment code
## <numeric> <list> <character>
## SAMEA112904734 NA TREATMENT,TRIAL,PEN CO
## SAMEA112904735 NA TREATMENT,TRIAL,PEN CC
## SAMEA112904737 NA TREATMENT,TRIAL,PEN CC
## SAMEA112904741 NA TREATMENT,TRIAL,PEN CC
## SAMEA112904746 NA TREATMENT,TRIAL,PEN CC
## SAMEA112904748 NA TREATMENT,TRIAL,PEN CO
## SAMEA112904749 NA TREATMENT,TRIAL,PEN CC
## SAMEA112904755 NA TREATMENT,TRIAL,PEN CC
## SAMEA112904762 NA TREATMENT,TRIAL,PEN CO
## SAMEA112904763 NA TREATMENT,TRIAL,PEN CC
## Treatment name Trial code Trial description Trial end
## <character> <character> <character> <character>
## SAMEA112904734 Probiotic CB Trial 2 2019-05-19
## SAMEA112904735 Control CC Trial 3 2019-07-14
## SAMEA112904737 Control CB Trial 2 2019-05-19
## SAMEA112904741 Control CC Trial 3 2019-07-14
## SAMEA112904746 Control CB Trial 2 2019-05-19
## SAMEA112904748 Probiotic CC Trial 3 2019-07-14
## SAMEA112904749 Control CA Trial 1 2019-03-11
## SAMEA112904755 Control CB Trial 2 2019-05-19
## SAMEA112904762 Probiotic CC Trial 3 2019-07-14
## SAMEA112904763 Control CA Trial 1 2019-03-11
## Trial start animal system canonical_url
## <character> <character> <character> <character>
## SAMEA112904734 2019-04-21 SAMEA112904734 chicken https://www.ebi.ac.u..
## SAMEA112904735 2019-06-09 SAMEA112904735 chicken https://www.ebi.ac.u..
## SAMEA112904737 2019-04-21 SAMEA112904737 chicken https://www.ebi.ac.u..
## SAMEA112904741 2019-06-09 SAMEA112904741 chicken https://www.ebi.ac.u..
## SAMEA112904746 2019-04-21 SAMEA112904746 chicken https://www.ebi.ac.u..
## SAMEA112904748 2019-06-09 SAMEA112904748 chicken https://www.ebi.ac.u..
## SAMEA112904749 2019-02-04 SAMEA112904749 chicken https://www.ebi.ac.u..
## SAMEA112904755 2019-04-21 SAMEA112904755 chicken https://www.ebi.ac.u..
## SAMEA112904762 2019-06-09 SAMEA112904762 chicken https://www.ebi.ac.u..
## SAMEA112904763 2019-02-04 SAMEA112904763 chicken https://www.ebi.ac.u..
## Average Daily Feed intake: Day 00 - 07
## <numeric>
## SAMEA112904734 19.39
## SAMEA112904735 22.35
## SAMEA112904737 18.18
## SAMEA112904741 21.68
## SAMEA112904746 19.75
## SAMEA112904748 21.10
## SAMEA112904749 18.67
## SAMEA112904755 23.36
## SAMEA112904762 21.15
## SAMEA112904763 19.67
## Average Daily Feed intake: Day 00 - 07, unit
## <character>
## SAMEA112904734 g
## SAMEA112904735 g
## SAMEA112904737 g
## SAMEA112904741 g
## SAMEA112904746 g
## SAMEA112904748 g
## SAMEA112904749 g
## SAMEA112904755 g
## SAMEA112904762 g
## SAMEA112904763 gThe sampleMap slot now contains mappings between each unique sample and the corresponding individual animal. There are as many rows as there are total samples.
The “colname” column refers to the samples in the omic dataset identified in the “assay” column, while the “primary” column provides information about the animals. You will notice that some animals are listed multiple times, reflecting the multiple omics and time points collected for those individuals.
sampleMap(mae)
## DataFrame with 33 rows and 3 columns
## assay primary colname
## <factor> <character> <character>
## 1 BIOGENIC AMINES SAMEA112904734 SAMEA112906114
## 2 BIOGENIC AMINES SAMEA112904734 SAMEA112906592
## 3 BIOGENIC AMINES SAMEA112904735 SAMEA112906338
## 4 BIOGENIC AMINES SAMEA112904735 SAMEA112906810
## 5 BIOGENIC AMINES SAMEA112904737 SAMEA112906123
## ... ... ... ...
## 29 FATTY ACIDS SAMEA112904755 SAMEA112906112
## 30 FATTY ACIDS SAMEA112904755 SAMEA112906590
## 31 FATTY ACIDS SAMEA112904762 SAMEA112906315
## 32 FATTY ACIDS SAMEA112904762 SAMEA112906787
## 33 FATTY ACIDS SAMEA112904763 SAMEA112905916For information have a look at the intro vignette of the MultiAssayExperiment package.
| Option | Rows (features) | Cols (samples) | Recommended |
|---|---|---|---|
| assays | match | match | Data transformations |
| altExp | free | match | Alternative experiments |
| MultiAssay | free | free (mapping) | Multi-omic experiments |
Multi-assay analyses, discussed in Chapter 19 and Chapter 20, can be facilitated by the multi-assay data containers, TreeSummarizedExperiment and MultiAssayExperiment. These are scalable and contain different types of data in a single container, making this framework particularly suited for multi-assay microbiome data incorporating different types of complementary data sources in a single, reproducible workflow. An alternative experiment can be stored in altExp slot of the SE data container. Alternatively, both experiments can be stored side-by-side in an MAE data container.