7 Data wrangling
This chapter introduces several essential techniques for preparing data for analysis. These techniques include splitting data, modifying data, and converting data to a data.frame. Additionally, it explains how to merge multiple SummarizedExperiment objects when needed. For a basic understanding of TreeSummarizedExperiment please refer to Chapter 3.
7.1 Splitting
You can split the data based on variables by using the functions agglomerateByRanks() and splitOn(). The former is detailed in Chapter 10.
If you want to split the data based on a variable other than taxonomic rank, use splitOn(). It works for row-wise and column-wise splitting. Splitting the data may be useful, for example, if you want to analyze data from different cohorts separately.
The following example demonstrates how to identify the most abundant taxonomic features for each sample type. Given that some samples originate from different origin, we expect differences between sample types. While getTop() can be used to determine the most abundant features across the entire dataset, this approach would obscure features that are specific to individual sample types.
library(mia)
library(knitr)
data("GlobalPatterns")
tse <- GlobalPatterns
# Agglomerate to phyla
tse <- agglomerateByRank(tse, rank = "Phylum")
# Split data based on sample type
tse_list <- splitOn(tse, by = "samples", group = "SampleType")
# Loop over the list of TreeSEs, and get top features for each
top_taxa <- sapply(tse_list, getTop)
top_taxa |> kable()| Soil | Feces | Skin | Tongue | Freshwater | Freshwater (creek) | Ocean | Sediment (estuary) | Mock |
|---|---|---|---|---|---|---|---|---|
| Proteobacteria | Firmicutes | Firmicutes | Proteobacteria | Cyanobacteria | Cyanobacteria | Proteobacteria | Proteobacteria | Firmicutes |
| Acidobacteria | Bacteroidetes | Proteobacteria | Firmicutes | Actinobacteria | Proteobacteria | Bacteroidetes | Bacteroidetes | Bacteroidetes |
| Verrucomicrobia | Cyanobacteria | Actinobacteria | Bacteroidetes | Proteobacteria | Bacteroidetes | Cyanobacteria | Cyanobacteria | Proteobacteria |
| Bacteroidetes | Actinobacteria | Bacteroidetes | Actinobacteria | Bacteroidetes | Verrucomicrobia | Actinobacteria | Crenarchaeota | Actinobacteria |
| Actinobacteria | Proteobacteria | Fusobacteria | Fusobacteria | Verrucomicrobia | Chloroflexi | Euryarchaeota | Planctomycetes | Tenericutes |
7.2 Add or modify variables
The information contained by the colData of a TreeSummarizedExperiment can be added and/or modified by accessing the desired variables. You might want to add or modify this data to include new variables or update existing ones, which can be essential for ensuring that all relevant metadata is available for subsequent analyses.
data("GlobalPatterns")
tse <- GlobalPatterns
# Modify the Description entries
colData(tse)$Description <- paste(
colData(tse)$Description, "modified description"
)
# View modified variable
tse$Description |> head()
## [1] "Calhoun South Carolina Pine soil, pH 4.9 modified description"
## [2] "Cedar Creek Minnesota, grassland, pH 6.1 modified description"
## [3] "Sevilleta new Mexico, desert scrub, pH 8.3 modified description"
## [4] "M3, Day 1, fecal swab, whole body study modified description"
## [5] "M1, Day 1, fecal swab, whole body study modified description"
## [6] "M3, Day 1, right palm, whole body study modified description"New information can be added to the experiment by creating a new variable.
Alternatively, you can add a whole table by merging it with existing colData.
Similar steps can also be applied to rowData. If you have an assay whose rows and columns align with the existing ones, you can add the assay easily to the TreeSummarizedExperiment object.
Here we add an assay that has random numbers but in real life these steps might come in handy after you have transformed the data with custom transformation that cannot be found from mia.
Now we can see that the TreeSummarizedExperiment object has an additional assay called “random”. When adding new samples or features to your existing dataset, you can use cbind() to combine columns for new features or rbind() to add rows for new samples.
tse2 <- cbind(tse, tse)
tse2
## class: TreeSummarizedExperiment
## dim: 19216 52
## metadata(0):
## assays(2): counts random
## rownames(19216): 549322 522457 ... 200359 271582
## rowData names(7): Kingdom Phylum ... Genus Species
## colnames(52): CL3 CC1 ... Even2 Even3
## colData names(10): X.SampleID Primer ... var1 var2
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):
## rowLinks: a LinkDataFrame (19216 rows)
## rowTree: 1 phylo tree(s) (19216 leaves)
## colLinks: NULL
## colTree: NULLHowever, the aforementioned functions assume that the rows align correctly when combining columns, and vice versa. In practice, this is often not the case; for example, samples may have different feature sets. In such situations, using a merging approach is the appropriate method.
7.3 Merge data
The mia package has a mergeSEs() function that merges multiple SummarizedExperimentobjects. For example, it is possible to combine multiple TreeSummarizedExperiment objects which each includes one sample.
mergeSEs() works much like standard joining operations. It combines rows and columns and allows you to specify the merging method.
# Take subsets for demonstration purposes
tse1 <- tse[, 1]
tse2 <- tse[, 2]
tse3 <- tse[, 3]
tse4 <- tse[1:100, 4]# With inner join, we want to include all shared rows. When using mergeSEs
# function all samples are always preserved.
tse <- mergeSEs(list(tse1, tse2, tse3, tse4), join = "inner")
tse
## class: TreeSummarizedExperiment
## dim: 100 4
## metadata(0):
## assays(1): counts
## rownames(100): 239672 243675 ... 104332 159421
## rowData names(7): Kingdom Phylum ... Genus Species
## colnames(4): CC1 CL3 M31Fcsw SV1
## colData names(10): X.SampleID Primer ... var1 var2
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):
## rowLinks: a LinkDataFrame (100 rows)
## rowTree: 1 phylo tree(s) (19216 leaves)
## colLinks: NULL
## colTree: NULL# Left join preserves all rows of the 1st object
tse <- mergeSEs(tse1, tse4, missing.values = 0, join = "left")
tse
## class: TreeSummarizedExperiment
## dim: 19216 2
## metadata(0):
## assays(1): counts
## rownames(19216): 239672 243675 ... 239967 254851
## rowData names(7): Kingdom Phylum ... Genus Species
## colnames(2): CL3 M31Fcsw
## colData names(10): X.SampleID Primer ... var1 var2
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):
## rowLinks: a LinkDataFrame (19216 rows)
## rowTree: 1 phylo tree(s) (19216 leaves)
## colLinks: NULL
## colTree: NULL7.4 Melting data
For several custom analysis and visualization packages, such as those from tidyverse, the SummarizedExperiment data can be converted to a long data.frame format with meltSE().
| FeatureID | SampleID | counts | Kingdom | Phylum | Class | Order | Family | Genus | Species | X.SampleID | Primer | Final_Barcode | Barcode_truncated_plus_T | Barcode_full_length | SampleType | Description | NewVariable | var1 | var2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 239672 | CL3 | 0 | Archaea | Crenarchaeota | C2 | B10 | NA | NA | NA | CL3 | ILBC_01 | AACGCA | TGCGTT | CTAGCGTGCGT | Soil | Calhoun South Carolina Pine soil, pH 4.9 modified description | 0.2809 | 0.6711 | 0.7963 |
| 239672 | M31Fcsw | 0 | Archaea | Crenarchaeota | C2 | B10 | NA | NA | NA | M31Fcsw | ILBC_04 | AAGAGA | TCTCTT | TCGACATCTCT | Feces | M3, Day 1, fecal swab, whole body study modified description | 0.0772 | 0.0900 | 0.4838 |
| 243675 | CL3 | 0 | Archaea | Crenarchaeota | C2 | B10 | NA | NA | NA | CL3 | ILBC_01 | AACGCA | TGCGTT | CTAGCGTGCGT | Soil | Calhoun South Carolina Pine soil, pH 4.9 modified description | 0.2809 | 0.6711 | 0.7963 |
| 243675 | M31Fcsw | 0 | Archaea | Crenarchaeota | C2 | B10 | NA | NA | NA | M31Fcsw | ILBC_04 | AAGAGA | TCTCTT | TCGACATCTCT | Feces | M3, Day 1, fecal swab, whole body study modified description | 0.0772 | 0.0900 | 0.4838 |
| 444679 | CL3 | 0 | Archaea | Crenarchaeota | C2 | B10 | NA | NA | NA | CL3 | ILBC_01 | AACGCA | TGCGTT | CTAGCGTGCGT | Soil | Calhoun South Carolina Pine soil, pH 4.9 modified description | 0.2809 | 0.6711 | 0.7963 |
| 444679 | M31Fcsw | 0 | Archaea | Crenarchaeota | C2 | B10 | NA | NA | NA | M31Fcsw | ILBC_04 | AAGAGA | TCTCTT | TCGACATCTCT | Feces | M3, Day 1, fecal swab, whole body study modified description | 0.0772 | 0.0900 | 0.4838 |
For MultiAssayExperiment data, you can instead utilize longForm().
| assay | primary | rowname | colname | value | Rat | Diet |
|---|---|---|---|---|---|---|
| microbiota | C1 | GAYR01026362.62.2014 | C1 | 0 | 1 | High-fat |
| microbiota | C1 | CVJT01000011.50.2173 | C1 | 1 | 1 | High-fat |
| microbiota | C1 | KF625183.1.1786 | C1 | 0 | 1 | High-fat |
| microbiota | C1 | AYSG01000002.292.2076 | C1 | 0 | 1 | High-fat |
| microbiota | C1 | CCPS01000022.154.1916 | C1 | 7 | 1 | High-fat |
| microbiota | C1 | KJ923794.1.1762 | C1 | 0 | 1 | High-fat |
7.5 Tidy R programming
The tidy paradigm, first introduced in the tidyverse, promotes an intuitive and easy-to-learn coding style, which has made it popular among users. tidyomics bridges the gap between Bioconductor’s SummarizedExperiment ecosystem and the tidyverse (Hutchison et al. 2024).
The package tidySingleCellExperiment can be used to manage data in SummarizedExperiment. For instance, we can easily view the object as a tibble abstraction, familiar from tidyverse.
data("GlobalPatterns")
tse <- GlobalPatterns
library(tidySingleCellExperiment)
tse
## # A SingleCellExperiment-tibble abstraction: 26 × 8
## # [90mFeatures=19216 | Cells=26 | Assays=counts[0m
## .cell X.SampleID Primer Final_Barcode Barcode_truncated_plus_T
## <chr> <fct> <fct> <fct> <fct>
## 1 CL3 CL3 ILBC_01 AACGCA TGCGTT
## 2 CC1 CC1 ILBC_02 AACTCG CGAGTT
## 3 SV1 SV1 ILBC_03 AACTGT ACAGTT
## 4 M31Fcsw M31Fcsw ILBC_04 AAGAGA TCTCTT
## 5 M11Fcsw M11Fcsw ILBC_05 AAGCTG CAGCTT
## 6 M31Plmr M31Plmr ILBC_07 AATCGT ACGATT
## # ℹ 20 more rows
## # ℹ 3 more variables: Barcode_full_length <fct>, SampleType <fct>, …
## rowLinks: a LinkDataFrame (19216 rows)
## rowTree: 1 phylo tree(s) (19216 leaves)
## colLinks: NULL
## colTree: NULLBy utilizing tidy programming, we could then effortlessly manipulate the data. For instance, we can calculate the mean library size in each sample type. This is done by first calculating library size for each sample and then summarizing total counts in each sample type; all done by using tidy paradigm. For more information on library size, see Chapter 8.
tse %>%
join_features(features = rownames(tse), shape = "long") %>%
group_by(.cell) %>%
mutate(total_counts = sum(.abundance_counts)) %>%
group_by(SampleType) %>%
summarise(total_counts = mean(total_counts))
## # A tibble: 9 × 2
## SampleType total_counts
## <fct> <dbl>
## 1 Feces 1442116
## 2 Freshwater 1667452
## 3 Freshwater (creek) 1741407.
## 4 Mock 1088484.
## 5 Ocean 1218451
## 6 Sediment (estuary) 277173.
## # ℹ 3 more rowsWe can then filter samples and leverage ggplot2 for plotting. In the example below, we select soil samples and visualize their abundances with a boxplot. Of course, these tidy commands can be combined with mia tools. Before plotting, we apply CLR transformation by using trasformAssay() (see more on transformation from Chapter 11).
library(ggplot2)
tse %>%
filter(SampleType == "Soil") %>%
transformAssay(method = "clr", pseudocount = TRUE) %>%
join_features(features = rownames(tse), shape = "long") %>%
ggplot(aes(x = .cell, y = .abundance_clr, fill = .cell)) +
geom_boxplot()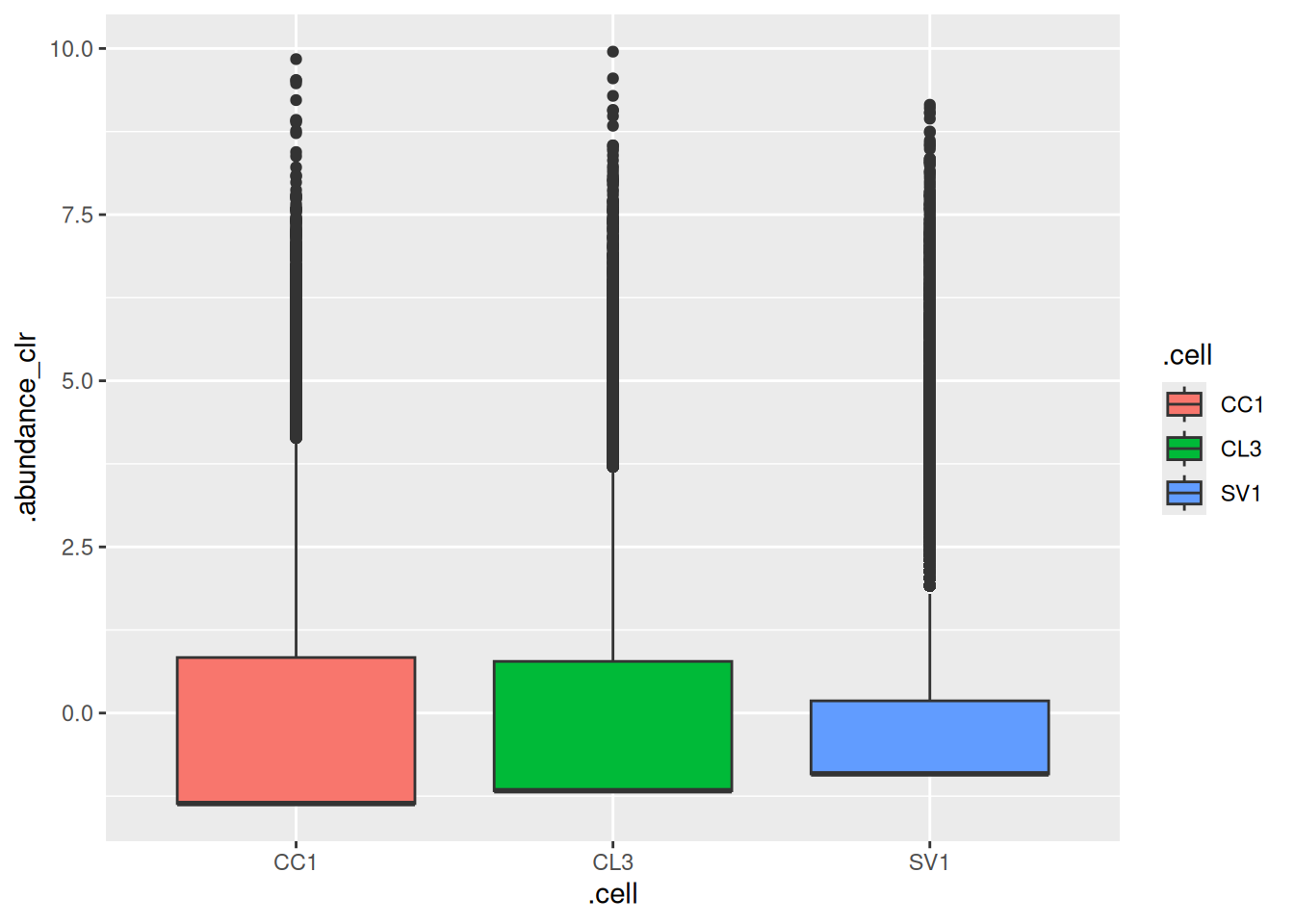
As demonstrated, tidy R programming can be effectively used to manage TreeSummarizedExperiment objects. Please refer to tidySummarizedExperiment and tidySingleCellExperiment vignettes for more examples.